Definition And Genetics
- Inherited metabolic disorders characterized by defects in the enzymes or transporters of the urea cycle.
- The metabolic block leads to the toxic accumulation of nitrogenous waste in the form of ammonia, accompanied by a deficiency of downstream intermediates such as arginine and citrulline.
- The collective incidence is approximately 1 in 35,000 live births.
- All disorders are inherited in an autosomal recessive manner, except Ornithine Transcarbamylase (OTC) deficiency, which is inherited as an X-linked recessive trait.
Classification And Etiology
The urea cycle requires five catalytic enzymes, a cofactor-synthesizing enzyme, and transport proteins.
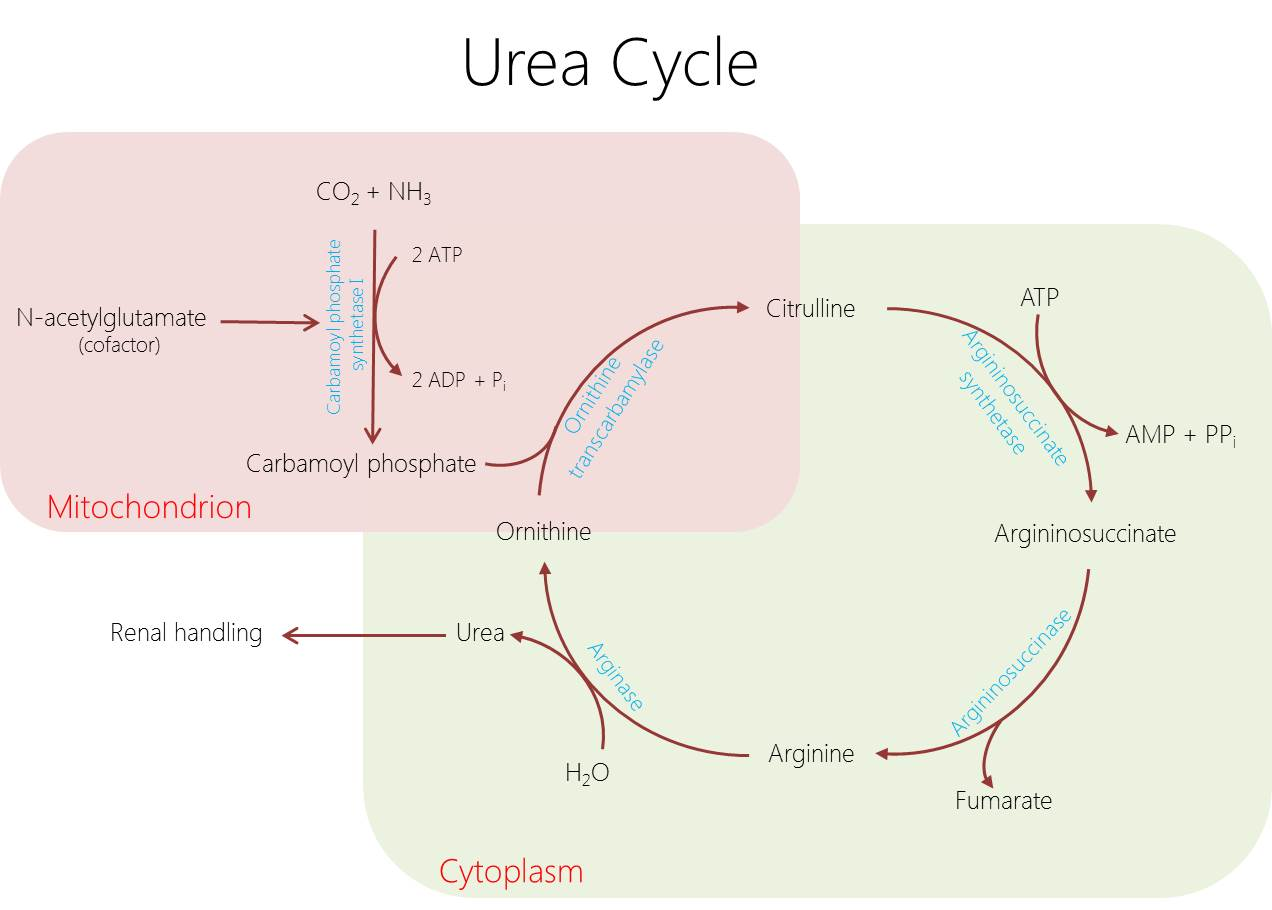
| Defect Location | Specific Enzyme Or Transporter | Associated Disorder |
|---|---|---|
| Proximal (Mitochondrial) | N-Acetylglutamate Synthase (NAGS) | NAGS deficiency (failure to activate CPS1) |
| Proximal (Mitochondrial) | Carbamoyl Phosphate Synthetase 1 (CPS1) | CPS1 deficiency (rate-limiting step) |
| Proximal (Mitochondrial) | Ornithine Transcarbamylase (OTC) | OTC deficiency |
| Distal (Cytosolic) | Argininosuccinate Synthetase (ASS) | Citrullinemia Type I |
| Distal (Cytosolic) | Argininosuccinate Lyase (ASL) | Argininosuccinic aciduria |
| Distal (Cytosolic) | Arginase 1 (ARG1) | Argininemia |
| Transporters | ORNT1 | Hyperornithinemia-Hyperammonemia-Homocitrullinemia (HHH) syndrome |
| Transporters | Citrin (SLC25A13) | Citrullinemia Type II |
Pathophysiology
- Hyperammonemia: Elevated ammonia crosses the blood-brain barrier, leading to astrocyte swelling and cerebral edema, mitochondrial dysfunction, and neurotransmitter dysregulation.
- Respiratory Alkalosis: Direct stimulation of the respiratory center by ammonia leads to central hyperventilation, a hallmark of these disorders.
- Amino Acid Imbalance: Depletion of urea cycle intermediates such as arginine and citrulline disrupts protein synthesis and nitric oxide production.
Clinical Features
Neonatal Onset (Severe Form)
- Infants are typically normal at birth but develop symptoms after a 24 to 48-hour symptom-free interval, coinciding with the loss of placental clearance and initiation of protein feeds.
- Neurological manifestations include poor feeding, lethargy, exaggerated startle response, hypotonia, progressing to seizures and deep coma.
- Systemic signs mimic neonatal sepsis, presenting with hypothermia and vomiting, alongside signs of cerebral edema such as a bulging fontanelle and abnormal posturing.
- Central hyperventilation causes characteristic respiratory alkalosis.
Late-Onset (Partial Defects)
- Presentations are episodic and triggered by catabolic stress (fever, fasting, surgery), postpartum stress, or high dietary protein loads.
- Symptoms include recurrent vomiting, ataxia, headaches, and lethargy.
- Psychiatric manifestations such as agitation, combativeness, hallucinations, and psychosis are common and frequently misdiagnosed.
- Patients often demonstrate voluntary protein aversion and may suffer from failure to thrive or intellectual disability.
Enzyme-Specific Phenotypes
- Arginase Deficiency: Uniquely presents with progressive spastic diplegia or tetraplegia, seizures, and intellectual disability, often without severe hyperammonemia.
- ASL Deficiency: Distinctly associated with trichorrhexis nodosa (brittle hair), hepatomegaly, and systemic hypertension.
Investigations
Diagnostic evaluation relies on identifying the exact biochemical blockage utilizing plasma and urine markers.
Screening And Initial Workup
| Investigation | Diagnostic Finding And Significance |
|---|---|
| Plasma Ammonia | Markedly elevated, often >150 µmol/L in neonates and >1000 µmol/L during acute crises. Must be transported on ice and analyzed immediately. |
| Arterial Blood Gas | Reveals respiratory alkalosis (high pH, low pCO2), distinguishing urea cycle disorders from organic acidemias which exhibit high anion gap metabolic acidosis. |
| Blood Glucose & LFTs | Typically normal, helping to exclude primary hypoglycemia or primary liver failure as the cause of encephalopathy. |
Biochemical Differentiation
| Suspected Disorder | Plasma Ammonia | Plasma Citrulline | Urine Orotic Acid | Key Additional Findings |
|---|---|---|---|---|
| CPS1 / NAGS Deficiency | High | Low/Absent | Low/Normal | High glutamine and alanine |
| OTC Deficiency | High | Low/Absent | High | High glutamine |
| Citrullinemia Type I (ASS) | High | Very High (>1000) | Normal/High | Low arginine |
| Argininosuccinic Aciduria (ASL) | High | Moderately High | Normal | High argininosuccinic acid |
| Argininemia (ARG1) | Mild/High | Normal | Normal | High arginine |
Confirmatory Tests: Molecular genetics via gene panel testing is the standard of care. Newborn screening detects citrullinemia and argininosuccinic aciduria but frequently misses OTC and CPS1 deficiencies.
Management
Acute Hyperammonemic Crisis (Emergency)
- Halt Nitrogen Intake: Immediately suspend all dietary protein intake for a maximum of 24 to 48 hours.
- Reverse Catabolism: Provide high-calorie intravenous infusions containing 10% Dextrose and Intralipid to achieve 100-120 kcal/kg, promoting anabolism.
- Nitrogen Scavenging Pharmacotherapy: Administer intravenous sodium benzoate (excreted as hippurate) and sodium phenylacetate or phenylbutyrate (excreted as phenylacetylglutamine) to provide alternate pathways for nitrogen disposal.
- Substrate Priming: Administer intravenous arginine (except in arginase deficiency) or citrulline to optimize residual urea cycle function.
- Specific Analog Therapy: Administer Carglumic Acid (Carbaglu) for patients with suspected or confirmed NAGS deficiency.
- Dialysis: Hemodialysis is strongly indicated if ammonia exceeds 500 µmol/L or fails to respond to medical management within 3 to 6 hours.
Chronic Maintenance
- Maintain a strict protein-restricted diet supplemented with essential amino acids to match growth demands.
- Administer chronic oral scavengers such as sodium phenylbutyrate or glycerol phenylbutyrate.
- Provide ongoing oral supplementation of L-citrulline or L-arginine.
- Liver transplantation is curative for primary hepatic enzyme defects (e.g., OTC, CPS1) and is indicated for poor metabolic control, though it cannot reverse pre-existing neurological damage.
Prognosis
- Mortality remains extremely high in severe neonatal-onset forms without rapid intervention.
- Morbidity, including intellectual disability, cerebral palsy, and epilepsy, strictly correlates with the severity and duration of the hyperammonemic coma.