Definition And Genetics
- Phenylketonuria is an autosomal recessive inborn error of amino acid metabolism.
- The disorder is caused by a deficiency of the hepatic enzyme Phenylalanine Hydroxylase (PAH).
- The genetic defect involves the PAH gene located on chromosome 12q23.2.
- Classic PKU accounts for >98% of cases, while 1-3% of cases result from defects in the synthesis or recycling of its cofactor, Tetrahydrobiopterin (BH4).
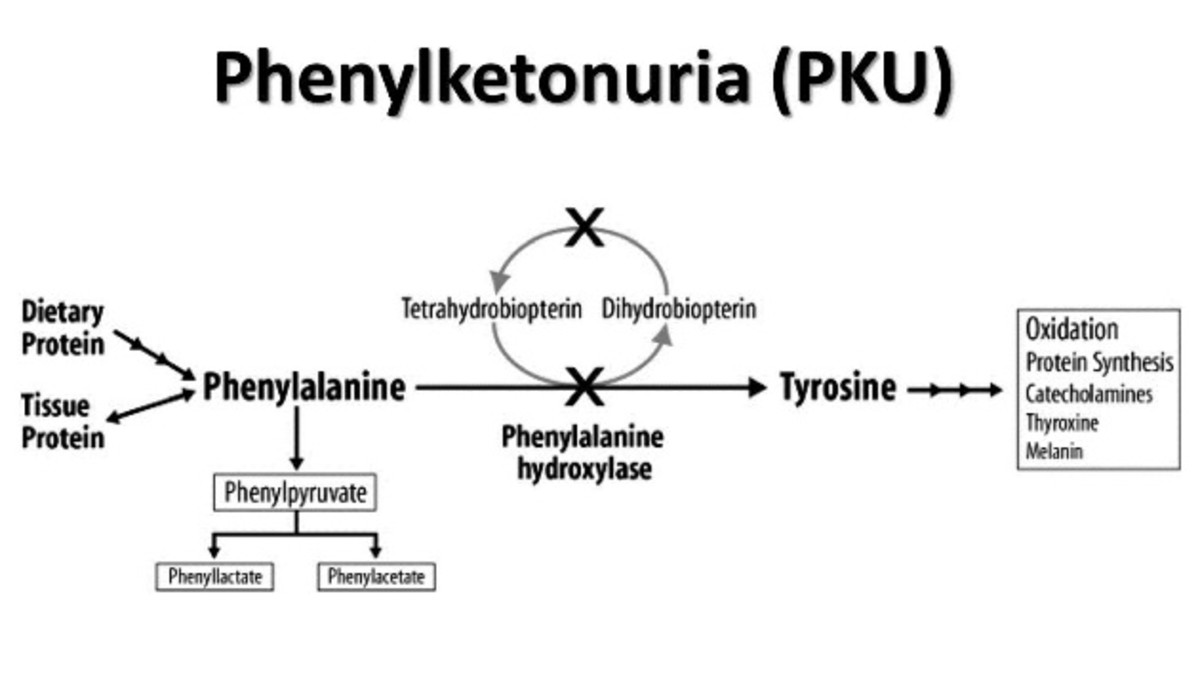
Pathophysiology
Metabolic Block And Neurotoxicity
- The deficiency of Phenylalanine Hydroxylase prevents the conversion of phenylalanine (Phe) to tyrosine.
- Excess phenylalanine is highly neurotoxic.
- Elevated phenylalanine saturates Large Neutral Amino Acid (LNAA) transporters at the blood-brain barrier, which competitively inhibits the brain uptake of other essential amino acids like tyrosine and tryptophan.
- This transport inhibition leads to severe central nervous system depletion of neurotransmitters (dopamine, serotonin, norepinephrine) and impaired cerebral protein synthesis.
Tyrosine Deficiency And Alternative Pathways
- Tyrosine becomes an essential amino acid, and its deficiency impairs the synthesis of melanin, epinephrine, and dopamine.
- Excess phenylalanine is diverted to alternative metabolic pathways, converting into phenylketones (phenylpyruvate, phenylacetate, and phenylacetylglutamine) which are excreted in the urine.
Clinical Classification
Classification is based on plasma phenylalanine levels on an unrestricted diet.
| Classification | Plasma Phenylalanine Levels |
|---|---|
| Classic PKU | >20 mg/dL (>1,200 µmol/L) |
| Mild PKU | 10–20 mg/dL (600–1,200 µmol/L) |
| Mild Hyperphenylalaninemia | 2–10 mg/dL (120–600 µmol/L) |
Clinical Features
Infants appear completely normal at birth due to effective placental clearance of phenylalanine. Without early intervention, severe symptoms develop.
Central Nervous System Manifestations
- Profound intellectual disability with an IQ <35 in 50-70% of untreated patients.
- Microcephaly and severe global developmental delay.
- Neurological signs include spasticity, hyperreflexia, tremors, and seizures (present in 25% of cases).
Behavioral And Dermatological Manifestations
- Behavioral issues include hyperactivity, aggressive tendencies, autistic features, and rhythmic rocking.
- Hypopigmentation is prominent, resulting in fair skin, blond hair, and blue eyes due to impoverished melanin synthesis.
- Patients frequently present with an eczematoid or seborrheic rash.
- A characteristic “mousey” or “musty” body and urine odor is present due to phenylacetic acid accumulation.
Diagnostic Investigations
Screening And Confirmatory Workup
| Investigation Type | Details and Findings |
|---|---|
| Newborn Screening (NBS) | Uses Tandem Mass Spectrometry (TMS) at 24–48 hours of life, targeting elevated Phe and increased Phe:Tyrosine ratio. |
| Plasma Amino Acids | Quantitative measurement reveals elevated phenylalanine and low or normal tyrosine. |
| Cofactor Screening | Mandatory for all hyperphenylalaninemia cases to rule out BH4 deficiency by measuring urine/blood pterins (neopterin, biopterin) and DHPR enzyme activity. |
| Molecular Genetics | PAH gene sequencing confirms the diagnosis in 98% of cases. |
| BH4 Loading Test | Oral sapropterin administration reduces Phe levels by >30% in BH4-responsive variants. |
Management
Dietary Therapy
- Dietary intervention is the standard of care and must be initiated immediately if phenylalanine levels exceed 6–10 mg/dL.
- Management requires lifelong restriction of natural protein to limit phenylalanine intake.
- Patients require specialized phenylalanine-free medical formulas supplemented with tyrosine, vitamins, and minerals.
- The therapeutic target is to maintain plasma phenylalanine levels strictly between 2–6 mg/dL (120–360 µmol/L) for life.
- Discontinuation of the diet in adulthood results in executive dysfunction and emotional lability.
Pharmacotherapy
- Sapropterin: A synthetic formulation of BH4 that is effective in BH4-responsive PKU variants, allowing for increased dietary protein tolerance.
- Pegvaliase: An injectable PEGylated phenylalanine ammonia-lyase enzyme substitution therapy for adults with uncontrolled levels, which converts phenylalanine to harmless metabolites.
- Large Neutral Amino Acids (LNAA): Given to competitively inhibit phenylalanine transport across the blood-brain barrier.
Maternal Phenylketonuria Syndrome
- High maternal phenylalanine levels are highly teratogenic to the developing fetus.
- Fetal consequences include microcephaly, intellectual disability, severe intrauterine growth restriction (IUGR), and congenital heart disease.
- Prevention mandates strict metabolic control (phenylalanine 2–6 mg/dL) prior to conception and maintained throughout the entire pregnancy.