Definition And Genetics
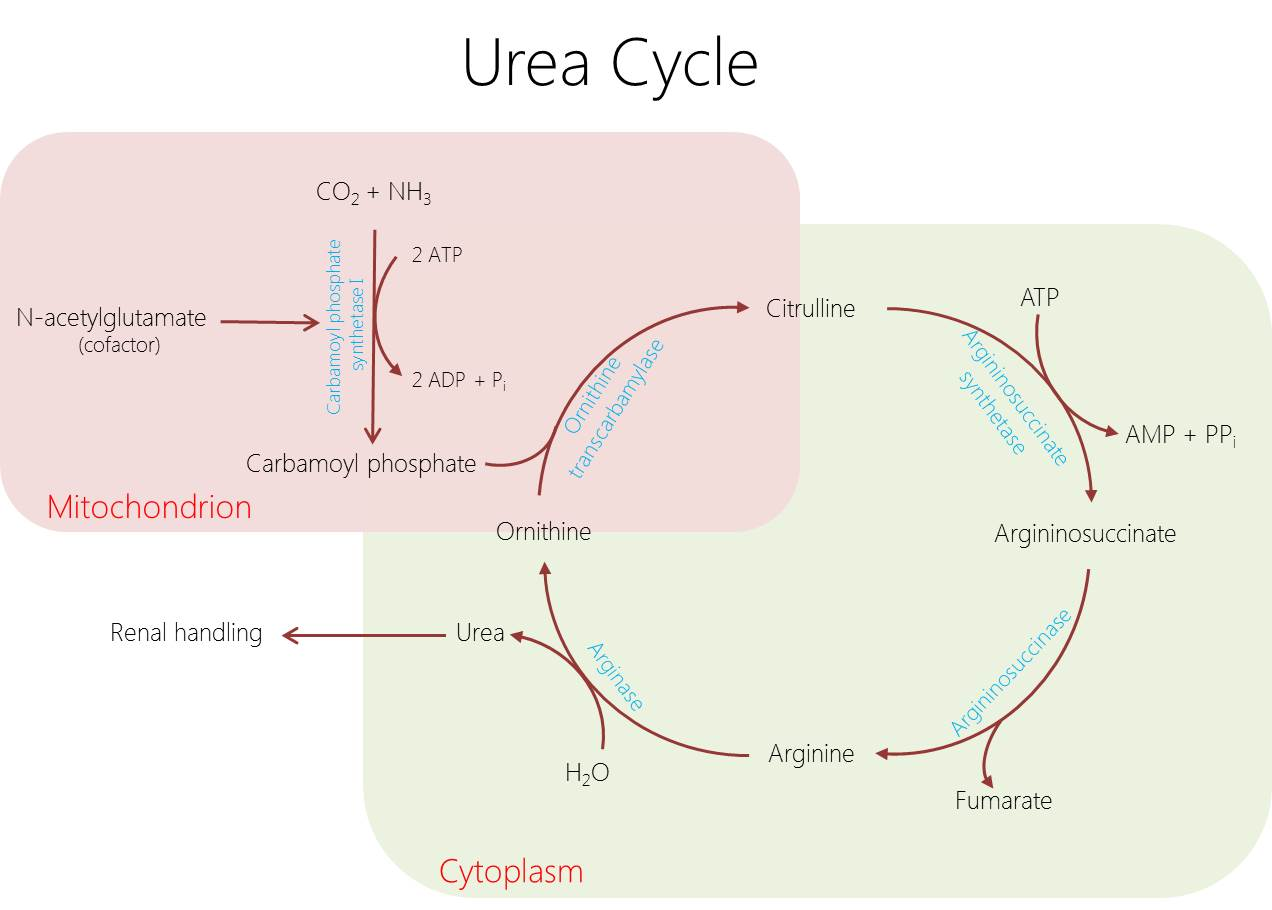
- The most common urea cycle disorder, characterized by the inability to convert ornithine and carbamoyl phosphate into citrulline.
- Inherited in an X-linked recessive manner, making it the only X-linked urea cycle disorder, while all others are autosomal recessive.
- Incidence ranges from approximately 1 in 14,000 to 1 in 77,000 live births.
- Caused by mutations in the OTC gene located at chromosome Xp11.4.
- Males present as hemizygotes with a severe, neonatal-onset phenotype, while heterozygous females have a variable phenotype ranging from asymptomatic to fatal due to skewed X-inactivation (Lyonization).
Pathophysiology
The primary defect is a deficiency of the hepatic mitochondrial enzyme ornithine transcarbamylase, which leads to distinct metabolic consequences.
| Metabolic Consequence | Mechanism And Effects |
|---|---|
| Hyperammonemia | Failure to detoxify ammonia into urea leads to severe neurotoxicity, cerebral edema, and astrocyte swelling. |
| Orotic Aciduria | Accumulated carbamoyl phosphate shunts into the pyrimidine synthesis pathway, generating excess orotic acid. |
| Amino Acid Imbalance | Depletion of downstream citrulline and arginine, rendering arginine an essential amino acid. |
Clinical Features
Neonatal Onset (Classic Severe Males)
- Infants typically appear healthy at birth but develop symptoms after 24–48 hours upon initiation of protein feeding.
- Presents as a sepsis mimic with poor feeding, lethargy, vomiting, and hypothermia.
- Neurologic manifestations include central hyperventilation secondary to cerebral edema, which causes a classic respiratory alkalosis.
- Rapidly progresses to somnolence, seizures, deep coma, and a bulging fontanelle from increased intracranial pressure.
Late-Onset (Partial Males And Heterozygous Females)
- Symptoms are often triggered by a high protein load, catabolic stress such as infection or surgery, postpartum stress, or valproate use.
- Patients may have a history of voluntary protein aversion, recurrent vomiting, and failure to thrive.
- Episodic presentations include ataxia, confusion, aggression, irritability, and psychiatric manifestations.
- Cyclical vomiting or migraine-like headaches are also common.
Investigations
Diagnostic workup relies on identifying the specific biochemical abnormalities and confirming the genetic defect.
| Investigation Type | Findings And Significance |
|---|---|
| Initial Screening | Plasma ammonia is markedly elevated (>150–200 µmol/L, often >1000 in neonates). Arterial blood gas shows respiratory alkalosis, which differentiates it from the metabolic acidosis seen in organic acidemias. |
| Plasma Amino Acids | Demonstrates low or absent citrulline, low arginine, and high glutamine (which acts as ammonia storage). |
| Urine Organic Acids | Markedly elevated orotic acid. This finding is critical to distinguish OTC deficiency from carbamoyl phosphate synthetase 1 deficiency, where orotic acid is low or normal. |
| Confirmatory Tests | Molecular genetics via OTC gene sequencing is the standard of care. Liver biopsy for enzyme assay is rarely needed. |
| Carrier Detection | An allopurinol challenge test can be used to detect carrier females by inducing orotic aciduria. |
Management
Acute Hyperammonemia Crisis (Medical Emergency)
- Stop nitrogen intake by suspending all natural dietary protein for a maximum of 24–48 hours.
- Reverse catabolism by administering high-calorie intravenous fluids (10–20% dextrose with lipids) to suppress endogenous protein breakdown.
- Initiate ammonia scavenging pharmacotherapy using intravenous sodium phenylacetate and sodium benzoate to conjugate glutamine and glycine for urinary excretion.
- Administer intravenous arginine or citrulline to prime the functional portions of the urea cycle and prevent catabolism.
- Hemodialysis or continuous renal replacement therapy is mandated if ammonia exceeds 500 µmol/L or if the patient fails medical therapy.
Chronic Maintenance
- Maintain a severely protein-restricted diet combined with essential amino acid supplements.
- Prescribe oral nitrogen scavengers, such as sodium benzoate, phenylbutyrate, or glycerol phenylbutyrate.
- Provide lifelong supplementation with L-citrulline or L-arginine.
- Liver transplantation is curative for the metabolic defect and is usually performed between 6 months and 1 year of age to prevent further neurocognitive damage, though it does not reverse pre-existing brain damage.
Prognosis
- Neonatal-onset disease carries high mortality, and survivors often suffer from significant developmental delay and intellectual disability, which is inversely proportional to the duration of the neonatal coma.
- Late-onset disease has a better prognosis if recognized and treated early, though the risk of hyperammonemia crises remains a lifelong threat.