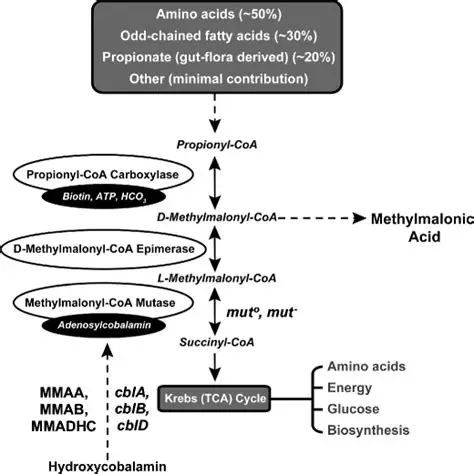
Autosomal recessive organic acidemia caused by a defect in the conversion of L-methylmalonyl-CoA to succinyl-CoA.
Classification Of Subtypes
Category
Subtypes And Characteristics
Isolated MMA
Mutase Deficiency:MMUT gene defects, classified into mut⁰ (complete deficiency) and mut⁻ (partial activity). Cobalamin Disorders: Defects in the synthesis of the adenosylcobalamin cofactor (cblA, cblB, cblD-MMA).
Toxic metabolites inhibit the glycine cleavage system.
Bone Marrow Suppression
Toxic effects lead to neutropenia and thrombocytopenia.
Clinical Features And Complications
Age Of Onset And Presentation
Neonatal Onset (Severe): Typically associated with mut⁰, presenting in the first week of life with a “sepsis-like” picture. Manifestations include poor feeding, vomiting, profound lethargy, and hypotonia. Patients show dehydration, hypothermia, Kussmaul breathing from severe acidosis, and rapid progression to seizures, coma, and death if untreated.
Chronic Or Intermittent (Late Onset): Associated with mut⁻ or cobalamin defects. Features include protein aversion, failure to thrive, chronic constipation, and developmental delay. Acute decompensation is typically triggered by catabolic stress such as infection, fasting, or high protein intake.
Long-Term Complications
System
Specific Complications
Neurologic
Metabolic stroke affecting the globus pallidus in the basal ganglia, leading to movement disorders like dystonia, chorea, and spasticity.
Renal
Chronic tubulointerstitial nephritis leading to progressive Chronic Kidney Disease, a feature unique to Methylmalonic Acidemia compared to other organic acidemias.
High anion gap metabolic acidosis, marked ketosis and ketonuria, mild to severe hyperammonemia, hyperglycinemia, and cytopenias (neutropenia, thrombocytopenia).
Urine Organic Acids
Massive elevation of Methylmalonic acid, alongside methylcitrate and 3-hydroxypropionate.
Plasma Acylcarnitine
Elevated C3 (propionylcarnitine) and C4DC (methylmalonylcarnitine).
Plasma Homocysteine
Essential to rule out combined Methylmalonic Acidemia and Homocystinuria (e.g., cblC disease).
Confirmatory Tests
Molecular genetics via gene panel testing for MMUT, MMAA, and MMAB. Clinical or in vitro Vitamin B12 responsiveness challenge.
Management
Acute Decompensation
Arrest Catabolism: Immediately halt exogenous protein intake for a maximum of 24-48 hours. Administer high-calorie intravenous fluids (10% Dextrose with intralipids) at 1.5 times maintenance to suppress endogenous protein breakdown.
Detoxification: Administer an intravenous loading dose of L-Carnitine (100 mg/kg) to facilitate the excretion of propionylcarnitine. Utilize ammonia scavengers such as Sodium Benzoate or Sodium Phenylacetate for hyperammonemia. Employ hemodialysis if severe acidosis or hyperammonemia is refractory to medical management.
Cofactor Therapy: Administer empirical intramuscular Hydroxocobalamin (Vitamin B12) at 1 mg/day.
Chronic Maintenance And Transplantation
Therapy Type
Interventions
Dietary
Low-protein diet restricting Isoleucine, Valine, Methionine, and Threonine, supplemented with specific medical foods. Avoidance of prolonged fasting.
Pharmacotherapy
Intramuscular Hydroxocobalamin for B12-responsive subtypes. Maintenance L-Carnitine (50-100 mg/kg/day). Gut sterilization using oral Metronidazole or Neomycin to decrease propionate production by enteric bacteria.
Transplantation
Liver transplantation corrects metabolic instability but fails to prevent chronic renal progression or reverse existing brain damage. Combined Liver-Kidney transplantation is indicated for patients exhibiting renal failure.
Prognosis
Prognosis heavily depends on the underlying subtype, with mut⁰ carrying the highest morbidity and poorest outcome.
Major long-term morbidities include irreversible intellectual disability, basal ganglia damage, and chronic renal failure.
While early diagnosis via Newborn Screening significantly improves initial survival, it does not completely prevent long-term systemic complications.