X-linked agammaglobulinemia (XLA), also known as Bruton agammaglobulinemia or infantile agammaglobulinemia, is a profound primary immunodeficiency resulting from a severe defect in B-lymphocyte development.
The disorder is transmitted as an X-linked trait, thereby predominantly affecting males, while female offspring of affected patients can carry the mutated gene.
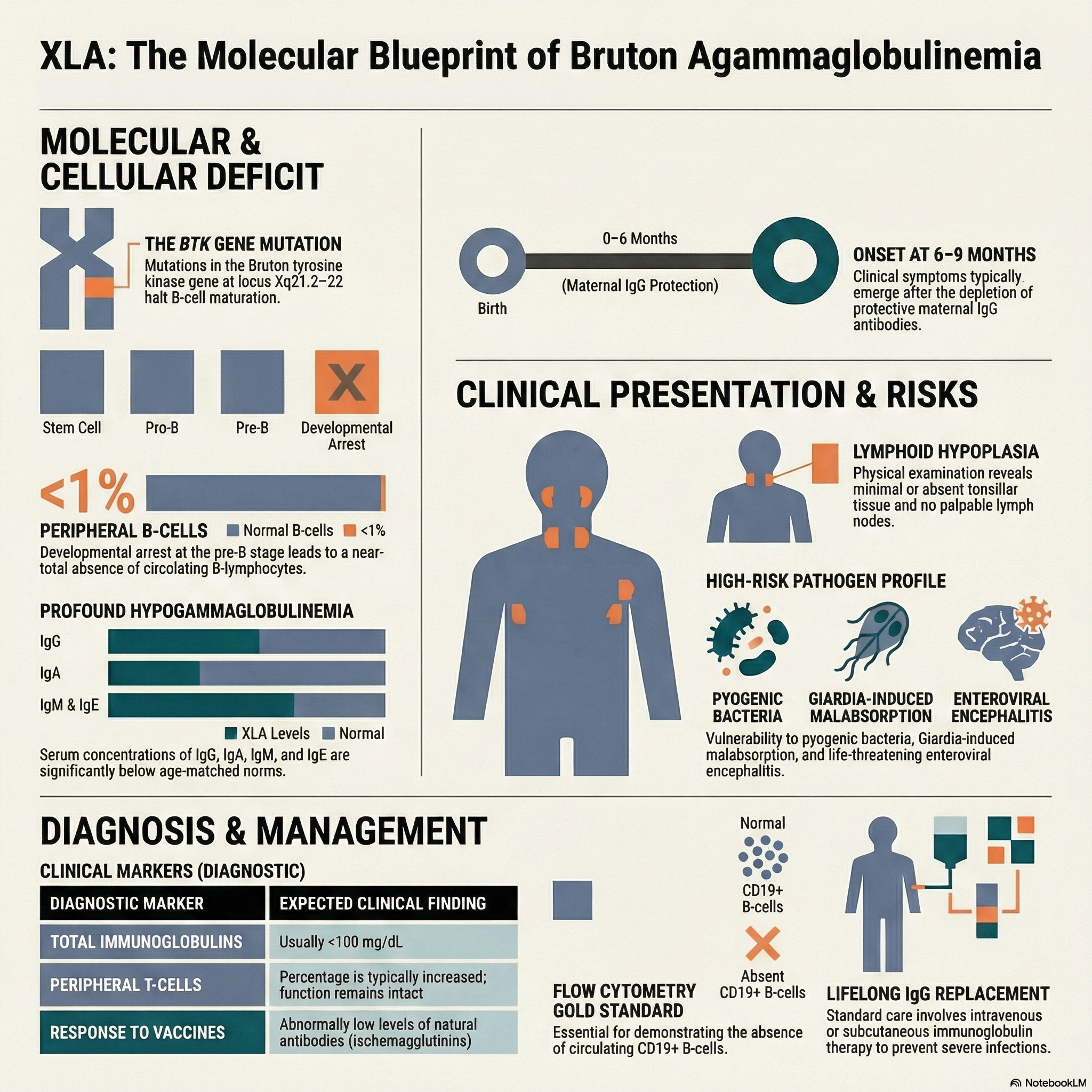
The condition is caused by a pathogenic variant in the gene encoding the B-cell protein tyrosine kinase (Btk), which maps to the q22 locus on the long arm of the X chromosome (Xq21.2–22).
The Btk protein is expressed in all B-lineage cells and plays a critical role in B-cell differentiation, maturation, and signaling.
Mutation in the BTK gene results in a developmental arrest of B cells, causing a profound absence of circulating B cells and severe hypogammaglobulinemia.
Although pre-B cells can be found in the bone marrow, the percentage of mature B lymphocytes in the peripheral blood is characteristically less than 1%.
Cell-mediated immunity is spared; the percentage of T cells is increased, the ratio of T-cell subsets is normal, T-cell function is completely intact, and the thymus appears anatomically normal.
Clinical Manifestations
Affected male infants typically remain asymptomatic for the first 6 to 9 months of life, as they are protected by maternally transmitted IgG antibodies acquired transplacentally.
Once maternal antibody levels wane, patients develop severe, recurrent infections caused primarily by extracellular pyogenic organisms, notably Streptococcus pneumoniae and Haemophilus influenzae.
The most common infectious presentations include sinusitis, otitis media, bronchitis, pneumonia, pyoderma, purulent conjunctivitis, sepsis, and meningitis.
Physical examination classically reveals marked lymphoid hypoplasia, characterized by small or completely absent tonsils and the absence of palpable lymph nodes.
Patients with XLA have a uniquely high susceptibility to chronic, frequently fatal central nervous system infections caused by enteroviruses, including echoviruses and coxsackieviruses.
An enterovirus-associated myositis that clinically resembles dermatomyositis has also been documented in these patients.
The administration of live attenuated viral vaccines, particularly the oral polio vaccine, can result in vaccine-associated paralytic poliomyelitis.
While most viral and fungal pathogens are handled normally, patients are prone to Mycoplasma and Ureaplasma urealyticum infections, which can cause severe purulent arthritis in large joints.
Gastrointestinal infections, particularly with Giardia lamblia, are common and can lead to chronic diarrhea, malabsorption, and weight loss.
Neutropenia is occasionally observed at the time of diagnosis during active infections, particularly those caused by staphylococcal species or Pseudomonas, which can lead to life-threatening invasive disease.
Laboratory Diagnosis
The diagnosis is highly suspected when total serum immunoglobulins are exceedingly low (typically <100 mg/dL), with specific concentrations of IgG, IgA, IgM, and IgE falling far below the 95% confidence limits for age-matched controls.
Patients lack specific functional antibodies; levels of natural isohemagglutinins (antibodies against type A and B red blood cell antigens) and antibodies generated in response to routine immunizations are abnormally low.
Flow cytometry is a mandatory diagnostic tool and demonstrates a virtual absence (less than 1%) of CD19+ B cells in the peripheral circulation.
The use of flow cytometry is critical to distinguish XLA from other humoral immunodeficiencies, such as common variable immunodeficiency (CVID), hyper-IgM syndrome, and transient hypogammaglobulinemia of infancy.
A definitive diagnosis is established through genetic testing to detect pathogenic variants in the BTK gene or the absence of the Btk protein.
Management and Complications
The cornerstone of medical management is lifelong immunoglobulin replacement therapy (administered intravenously or subcutaneously) to provide passive immunity and prevent recurrent infections.
Vigilant monitoring and the aggressive use of appropriate prophylactic or therapeutic antibiotics are essential to manage documented infections and decrease morbidity.
Despite adequate immunoglobulin replacement therapy, some patients continue to experience recurrent infections, and chronic respiratory complications such as severe bronchiectasis and chronic obstructive lung disease may persist.
Gastrointestinal disease is an increasingly recognized complication, and some patients may develop a chronic colitis that clinically mimics inflammatory bowel disease.