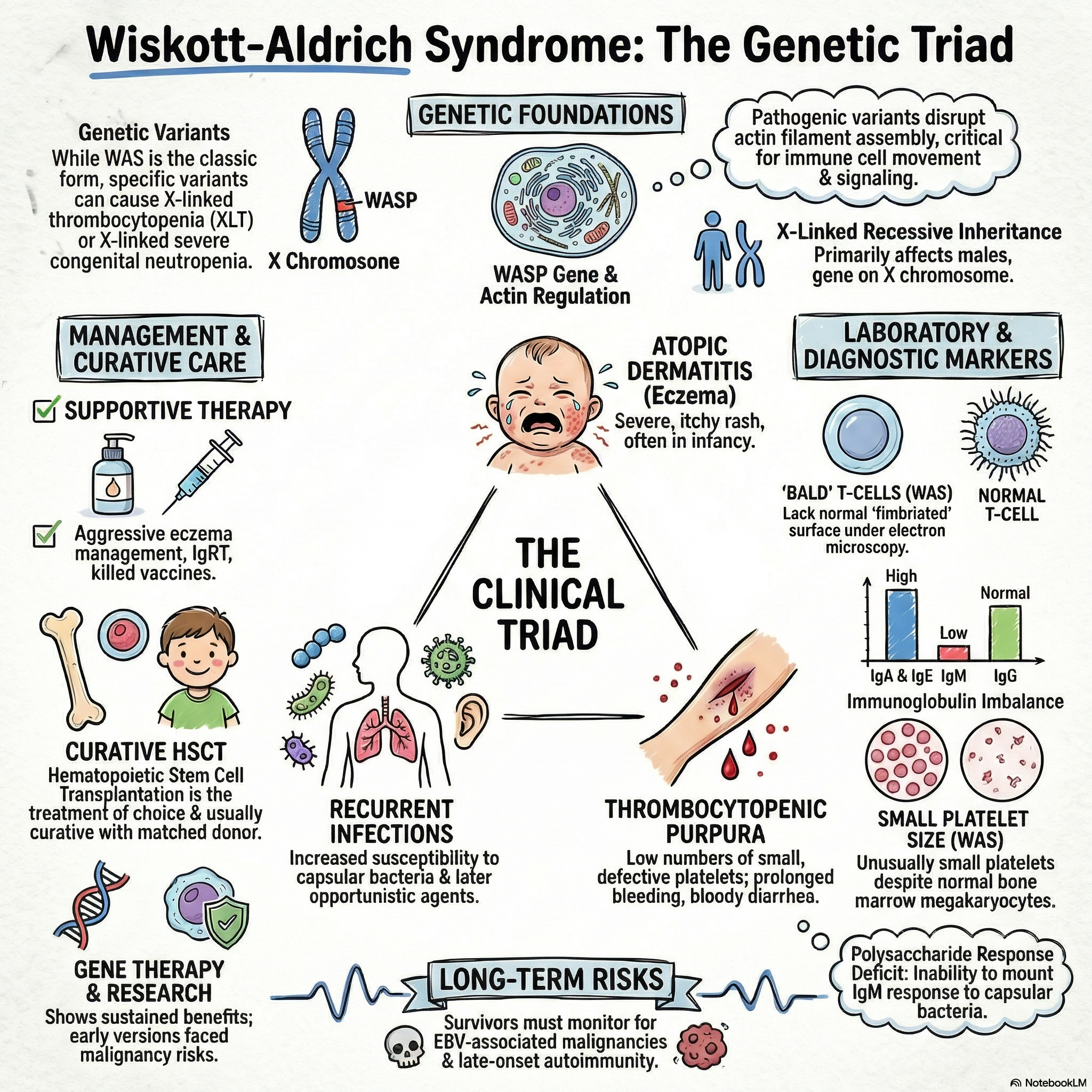
Wiskott-Aldrich syndrome is a combined immunodeficiency inherited as an X-linked recessive disorder.
The disease is caused by pathogenic variants in the WASP gene, which encodes the Wiskott-Aldrich syndrome protein.
The Wiskott-Aldrich syndrome protein is responsible for controlling the assembly of actin filaments, a process critically required for normal cell migration and cell-cell interactions.
The underlying immunologic defect appears to stem from the inability of T cells to provide adequate help to B cells.
Under electron microscopy, the T cells of affected patients uniquely lack the markedly fimbriated surface characteristic of normal T cells.
Clinical Manifestations
The classic clinical presentation of Wiskott-Aldrich syndrome consists of a triad of atopic dermatitis (eczema), congenital thrombocytopenia, and an increased susceptibility to infections.
Bleeding manifestations typically present very early in infancy, frequently presenting as prolonged bleeding from the circumcision site or as bloody diarrhea.
Patients are highly susceptible to recurrent pyogenic infections, such as otitis media, pneumonia, meningitis, and sepsis, which are primarily driven by encapsulated bacteria like Streptococcus pneumoniae and Haemophilus influenzae.
Later in the disease course, patients experience a higher frequency of infections with opportunistic organisms like Pneumocystis jirovecii and viruses from the herpesvirus family.
Affected individuals have a significant risk of developing autoimmune conditions, particularly autoimmune hemolytic anemia.
There is a marked predisposition to developing malignancies, notably Epstein-Barr virus (EBV)-associated malignancies and lymphoreticular cancers.
Physical examination may also reveal hepatosplenomegaly and lymphadenopathy.
Laboratory Diagnosis
A complete blood count reveals the hallmark finding of thrombocytopenia associated with characteristically small, defective platelets.
Peripheral blood eosinophilia is a common laboratory finding.
The classic serum immunoglobulin profile demonstrates a low level of IgM, elevated levels of IgA and IgE, and a normal or slightly decreased level of IgG.
Patients exhibit a profound impairment in their humoral immune response to polysaccharide antigens, which is evidenced by absent or greatly diminished isohemagglutinins and poor antibody responses following immunization with polysaccharide vaccines.
Evaluation of cellular immunity typically shows lymphopenia, moderately reduced percentages of T cells, and variably depressed lymphocyte proliferative responses to mitogen stimulation.
Management and Treatment
General supportive care involves appropriate nutrition and the aggressive management of eczema and any associated cutaneous infections.
Because of their profound antibody deficiency, patients require lifelong immunoglobulin replacement therapy, regardless of their specific serum immunoglobulin isotype levels.
Antimicrobial prophylaxis against Pneumocystis jirovecii and herpes simplex virus is frequently recommended to prevent life-threatening opportunistic infections.
Patients should only be administered killed vaccines, as live vaccines pose a severe infection risk in this immunocompromised state.
Splenectomy may become necessary for patients experiencing severe, refractory thrombocytopenia; however, these patients will subsequently require lifelong penicillin prophylaxis to protect against encapsulated organisms.
Hematopoietic stem cell transplantation (HSCT) is the definitive treatment of choice and is usually curative when a high-quality matched donor is available.
Ex vivo gene transfer to autologous hematopoietic stem cells has resulted in sustained benefits in several patients, though early trials of gene therapy were complicated by insertional mutagenesis leading to malignancy.