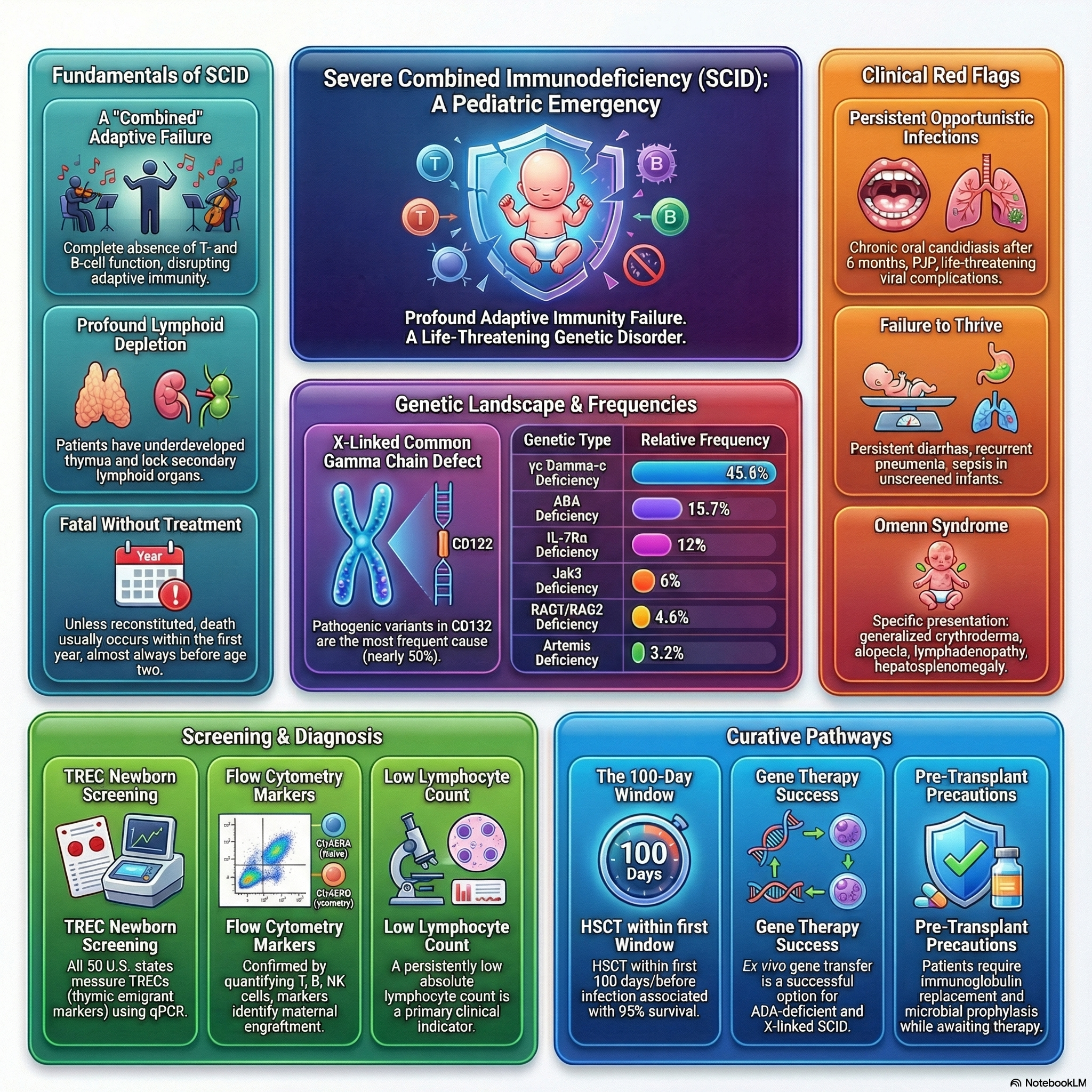
Severe combined immunodeficiency (SCID) represents the most severe form of primary immune deficiency, caused by diverse pathogenic gene variants that lead to the profound absence of T-cell and B-cell function.
The fundamental defect disrupts lymphoid cell development, resulting in very small thymuses that are devoid of thymocytes, lack corticomedullary distinction, and lack Hassall’s corpuscles.
Peripheral lymphoid structures are also severely affected; the follicular and paracortical areas of the spleen are depleted of lymphocytes, and lymph nodes, tonsils, adenoids, and Peyer patches are either completely absent or extremely underdeveloped.
Without immunologic reconstitution, SCID is considered a true pediatric immunologic emergency and is almost invariably fatal within the first 1 to 2 years of life.
Genetic Classification and Phenotypes
SCID exhibits significant genetic heterogeneity and is primarily categorized based on the presence or absence of specific lymphocyte populations (T cells, B cells, and Natural Killer [NK] cells).
T- B+ SCID (T-cell absent, B-cell present):
The most common form is X-linked SCID, caused by pathogenic variants in the IL2RG gene (CD132) encoding the common gamma chain (γc) of cytokine receptors (IL-2, IL-4, IL-7, IL-9, IL-15, and IL-21).
Patients with X-linked SCID typically present with absent T and NK cells but normal or elevated numbers of B cells (T- B+ NK-).
Pathogenic variants in JAK3, which signals downstream of the common gamma chain, cause an autosomal recessive form of SCID that presents with an identical lymphocyte phenotype (T- B+ NK-) affecting both males and females.
IL-7Rα deficiency, CD45 deficiency, and CD3 deficiencies (arrest of thymocyte differentiation) also cause T- B+ forms of SCID.
T- B- SCID (Both T-cell and B-cell absent):
Autosomal recessive RAG1 and RAG2 deficiencies result in defective V(D)J recombination, leading to the absence of mature T and B cells.
Adenosine deaminase (ADA) deficiency causes an accumulation of toxic purine nucleosides leading to lymphocyte death, frequently accompanied by non-immunologic features such as pulmonary alveolar proteinosis, chondro-osseous dysplasia, and neurologic, hepatic, or renal abnormalities.
Defects in nonhomologous end joining (NHEJ) and DNA repair enzymes, such as Artemis deficiency, DNA-PK deficiency, and DNA ligase IV deficiency, cause T- B- SCID often associated with marked radiation sensitivity, microcephaly, and growth delay.
Reticular dysgenesis, caused by variants in adenylate kinase 2 (AK2), impairs mitochondrial energy metabolism and presents with severe combined immunodeficiency, profound neutropenia, and sensorineural deafness.
Clinical Manifestations
If not detected by newborn screening, infants with SCID most often present with severe, persistent infections during early infancy.
Common infectious presentations include chronic diarrhea, recurrent pneumonia, persistent otitis media, bacterial sepsis, and severe cutaneous infections.
Patients demonstrate extreme susceptibility to opportunistic pathogens, classically presenting with severe oral thrush from Candida albicans, pneumonia from Pneumocystis jirovecii (PJP), and severe infections from parainfluenza 3 virus, adenovirus, respiratory syncytial virus (RSV), cytomegalovirus (CMV), and Epstein-Barr virus (EBV).
Exposure to live-attenuated vaccines—such as rotavirus, measles-mumps-rubella-varicella (MMRV), oral poliovirus (OPV), or the bacille Calmette-Guérin (BCG) vaccine—frequently results in life-threatening, disseminated vaccine-strain infections.
Maternal Engraftment: Infants with SCID cannot reject foreign tissues and are highly susceptible to severe graft-versus-host disease (GVHD) caused by the transplacental passage of maternal T cells during pregnancy.
Maternal engraftment GVHD manifests with a generalized rash, hepatosplenomegaly, diarrhea, and expansion of allogeneic cells.
Omenn Syndrome: Caused by hypomorphic pathogenic variants in SCID-associated genes, allowing the generation of a few oligoclonal T cells that expand in an unregulated manner, mimicking GVHD.
Omenn syndrome is classically characterized by severe generalized erythroderma, desquamation, alopecia, lymphadenopathy, hepatosplenomegaly, striking eosinophilia, and elevated serum IgE levels.
Newborn Screening
Newborn screening allows for the early detection and treatment of SCID prior to the onset of severe infections, which has dramatically improved survival rates.
The standard screening assay utilizes a quantitative polymerase chain reaction (PCR) to measure T-cell receptor excision circles (TRECs) from dried blood spots.
TRECs are episomal DNA byproducts formed during the V(D)J rearrangement of T-cell receptor genes; they do not replicate during cell division and serve as a highly accurate biomarker for enumerating recent thymic emigrants.
A low or absent TREC count raises high suspicion for SCID and mandates immediate confirmatory immunologic evaluation.
Some newborn screening programs additionally assay kappa excision circles (KRECs) to simultaneously identify severe B-cell defects like agammaglobulinemia.
Laboratory Diagnosis
A complete blood count (CBC) typically reveals persistent and profound lymphopenia, defined as an absolute lymphocyte count (ALC) of less than 3,000 cells/µL in infants younger than 12 months.
A normal ALC does not reliably rule out SCID, as the uncontrolled proliferation of maternal T cells, autologous B cells, or NK cells can mask an underlying T-cell lymphopenia.
Flow cytometry is mandatory to precisely quantitate lymphocyte subsets, including T cells (CD3, CD4, CD8), B cells (CD19, CD20), and NK cells (CD16, CD56).
Flow cytometric analysis of CD45 isoforms helps differentiate naive T cells (CD45RA) from memory T cells (CD45RO); a predominance of memory T cells strongly suggests maternal engraftment or Omenn syndrome.
Functional T-cell evaluation is conducted through lymphocyte proliferation assays, where a proliferative response to the mitogen phytohemagglutinin (PHA) of less than 10% of a normal control confirms severe combined immunodeficiency.
Fluorescence in situ hybridization (FISH) for X and Y chromosomes can be utilized in male infants to confirm the presence of engrafted maternal (XX) T cells.
Definitive diagnosis requires targeted gene sequencing using a SCID or primary immunodeficiency gene panel to identify the specific pathogenic variant, which is critical for guiding conditioning regimens and evaluating gene therapy options.
Management and Treatment
SCID necessitates immediate intervention, placing the infant in strict isolation to limit exposure to infectious agents.
Supportive therapy must be initiated promptly, including regular immunoglobulin replacement (IVIG or SCIG) and initiation of prophylactic antimicrobials targeting PJP, viral, and fungal pathogens.
Administration of any live-attenuated viral or bacterial vaccines (such as BCG, OPV, MMR, or varicella) is strictly contraindicated due to the high risk of fatal disseminated disease.
To prevent fatal transfusion-associated GVHD from viable donor lymphocytes, all administered blood products must be exclusively irradiated or frozen.
Breastfeeding should be withheld until the CMV and EBV statuses of both the mother and the infant are established, to prevent transmission of these viruses through breast milk.
Hematopoietic Stem Cell Transplantation (HSCT): Allogeneic HSCT remains the most effective, definitive, and curative treatment for SCID.
Survival rates approach 95% when HSCT is performed optimally within the first 100 days of life, prior to the onset of systemic infections.
Transplantation utilizing an HLA-identical sibling donor is preferred; however, haploidentical parental grafts utilizing rigorous T-cell depletion techniques are frequently and successfully employed without the need for pre-transplant myeloablative chemotherapy in some subsets.
Gene Therapy: Ex vivo gene transfer using lentiviral vectors has demonstrated significant success for patients with X-linked SCID and ADA-deficient SCID.
The transition from retroviral to lentiviral vectors has successfully minimized the risk of insertional mutagenesis and subsequent leukemic-like clonal T-cell proliferation.
Enzyme Replacement Therapy: For patients with ADA-SCID, regular intramuscular injections of polyethylene glycol-modified adenosine deaminase (PEG-ADA) provide a temporary bridge to definitive treatment, although it does not yield immune reconstitution equivalent to HSCT or gene therapy.