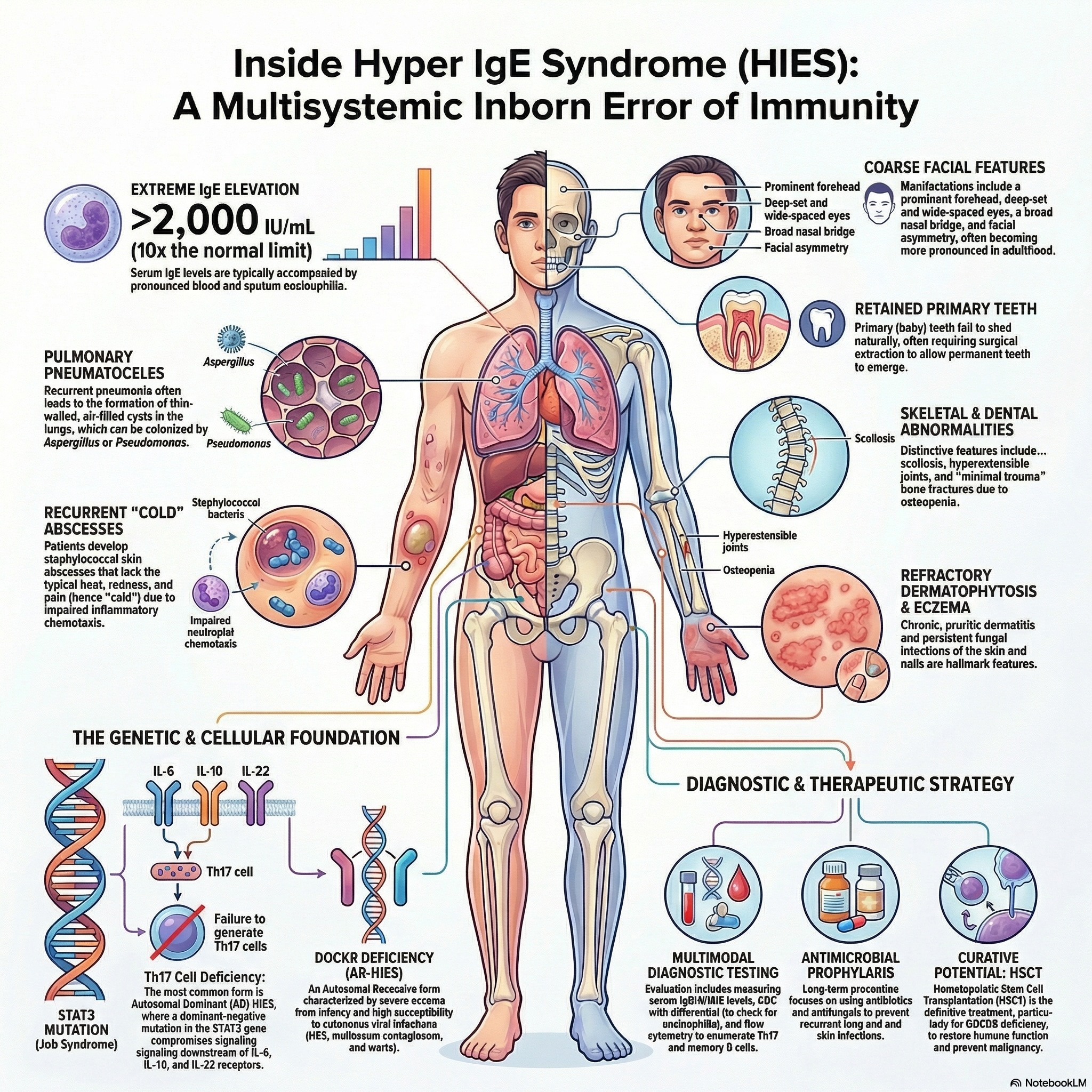
Hyper-IgE syndrome is a primary immunodeficiency with both autosomal dominant (AD) and autosomal recessive (AR) inheritance patterns.
Autosomal Dominant HIES (Job Syndrome): Caused by heterozygous pathogenic variants in the STAT3 gene, which act via a dominant negative effect.
STAT3 pathogenic variants compromise signaling pathways downstream of multiple cytokine receptors (IL-6, IL-10, IL-22), leading to a profound deficiency in T-helper type 17 (Th17) cells.
The immunologic defect impairs neutrophil chemotaxis and reduces macrophage microbicidal ability due to a failure of helper T-cells to produce interferon-gamma. This skews the immune system toward a Th2 response, triggering massive IgE and histamine overproduction, which blunts classic inflammatory reactions.
Autosomal Recessive HIES: Most commonly caused by pathogenic variants in the DOCK8 gene, resulting in T-cell lymphopenia, poor T-cell proliferation, and reduced numbers of regulatory T cells (Tregs) and natural killer (NK) cells.
Clinical Manifestations
Cutaneous: Patients with AD STAT3 deficiency classically present with early-onset, severe, pruritic eczema and recurrent Staphylococcus aureus skin abscesses. Because these abscesses lack the typical warmth, erythema, and tenderness of acute inflammation, they are uniquely termed “cold” abscesses.
Respiratory: Recurrent bacterial pneumonias frequently result in the formation of large, persistent lung pneumatoceles (cavities). These damaged areas are highly susceptible to secondary opportunistic colonization by Aspergillus species and Pneumocystis jirovecii.
Other Infections: Chronic mucocutaneous candidiasis occurs in over 70% of patients. Patients with the AR DOCK8 variant exhibit a profound susceptibility to severe, disseminated cutaneous viral infections, including herpes simplex, varicella, molluscum contagiosum, and human papillomavirus.
Facies (STAT3 deficiency): Characteristic facial dysmorphism includes a prominent forehead, deep-set and wide-spaced eyes, a broad nasal bridge, a wide and fleshy nasal tip, and facial asymmetry.
Skeletal and Connective Tissue (STAT3 deficiency): Non-immunologic features include delayed shedding of primary teeth, joint hyperextensibility, severe scoliosis, and osteopenia leading to recurrent minimal-trauma fractures. Vascular complications such as coronary and cerebral aneurysms may also occur.
Laboratory Diagnosis
A hallmark finding is an exceptionally high total serum IgE concentration, typically exceeding 2,000 IU/mL, though levels can fluctuate and may occasionally decline in adulthood.
Serum concentrations of IgG, IgA, and IgM are usually within normal limits.
Marked eosinophilia is consistently observed in the peripheral blood, sputum, and within histologic sections of affected tissues.
Flow cytometry generally shows normal absolute percentages of T, B, and NK lymphocytes, but reveals a specific and characteristic absence or severe deficiency of Th17 cells and a decreased percentage of memory T cells (CD45RO).
In vitro functional testing demonstrates normal T-lymphocyte proliferative responses to mitogens, but a very low or completely absent response to specific recall antigens.
Management
Therapy is predominantly supportive and aggressively directed at the prevention and early treatment of bacterial and fungal infections.
Continuous antimicrobial prophylaxis with trimethoprim-sulfamethoxazole is recommended to prevent recurrent Staphylococcus aureus and Streptococcus pneumoniae infections.
Antifungal prophylaxis with systemic agents, such as itraconazole, is routinely utilized to prevent invasive Aspergillus and Candida infections.
Immunoglobulin replacement therapy may be a beneficial adjunctive treatment to assist in infection prevention for selected patients.
For patients with AR DOCK8 deficiency, allogeneic hematopoietic stem cell transplantation (HSCT) is the definitive and curative treatment of choice, routinely performed early in life to prevent severe long-term complications and malignancy.