Components Of Hemostatic Process
Integral Components Of Hemostasis
Hemostasis requires complex integration of three major components: endothelial cells, platelets, and plasma coagulation factors.
Endothelial Cells
Form primary barrier against hemorrhage and thrombosis. Secrete specific substances modulating coagulation and vascular tone.
| Function | Secreted Substances |
|---|---|
| Repel platelets | Prostaglandin I2, adenosine diphosphate (ADP), nitric oxide |
| Initiate coagulation | Collagen, fibronectin |
| Promote platelet adhesion | Von Willebrand factor (vWF) |
| Promote fibrin dissolution | Tissue plasminogen activator (t-PA) |
| Inhibit thrombin | Heparin, thrombomodulin |
| Inhibit fibrin dissolution | t-PA inhibitor |
Platelets
Initiate primary hemostatic mechanism. Form temporary platelet plug. Adhere to areas of vascular injury. Activate and release intracellular contents.
- Dense Bodies Release: ADP, serotonin, calcium.
- Alpha Granules Release: Fibrinogen, vWF, factor V, high-molecular-weight kininogen, fibronectin, α-1-antitrypsin, platelet factor 4, platelet-derived growth factor.
- Additional Functions: Provide reaction surfaces for assembly of coagulation factors (VIIIa/Ca2+/IXa and Va/Ca2+/Xa complexes). Mediate blood vessel constriction via serotonin. Neutralize heparin.
Plasma Coagulation Factors
Synthesized primarily in liver. Factor VIII additionally produced by endothelial cells.
- Vitamin K-Dependent Factors: II, VII, IX, X require vitamin K for posttranslational gamma-carboxylation.
- Function: Circulate in zymogen form. Activate on platelet phospholipid surfaces. Generate thrombin from prothrombin.
Phases Of Hemostasis
Primary Hemostasis (Platelet Phase)
Endothelial injury exposes vWF and collagen from subendothelial matrix. Plasma vWF binds exposed collagen. Interacts with platelet GPIb receptor, tethering platelets. Platelet collagen receptors GPVI and α2β1 bind collagen. Platelets adhere, become activated. Conformational change occurs in αIIbβ3 receptor, enhancing avidity for vWF and fibrinogen. Mediates platelet-to-platelet interactions. Forms reversible aggregate platelet plug.
Secondary Hemostasis (Fibrin Thrombus Formation)
Divided into three overlapping phases.
Initiation Phase
Cell-based expression of tissue factor (TF) occurs at endothelial injury site. Factor VII binds exposed TF, becomes rapidly activated. FVIIa/TF complex generates FXa and FIXa. FXa activates FV. Complexes with FXa, generating small amounts of thrombin.
Amplification Phase
Procoagulant stimulus transfers to platelet surface at injury site. Small amounts of thrombin enhance platelet adhesion. Fully activate platelets. Activate FV, FVIII, FXI.
Propagation Phase
Tenase complex (FIXa+FVIIIa) assembles on platelet surface, generates FXa. Prothrombinase complex (FXa+FVa) assembles on platelet surface, generates massive thrombin burst. Thrombin activates FXIII. Cleaves fibrinopeptides A and B from fibrinogen. Residual peptide chains aggregate via hydrogen bonds forming fibrin monomers. Fibrin monomers convert into fibrin polymers via FXIIIa. Forms stable cross-linked fibrin network.
Fibrinolysis
Removes physiologically deposited fibrin. Plasminogen converts to enzymatically active plasmin. t-PA serves as principal intravascular activator. Urokinase-type plasminogen activator (u-PA) activates plasminogen independent of fibrin. Plasmin splits fibrin and fibrinogen into fibrin-degradation products (fragments X, Y, D, E; measured clinically as D-dimers).
Natural Inhibitors Of Coagulation
Regulate extension of clotting process to prevent systemic thrombosis.
| Inhibitor | Primary Function / Target Substrates |
|---|---|
| Antithrombin (AT) | Serine protease inhibitor; neutralizes thrombin, FIXa, FXa, FXIa, FXIIa. |
| Tissue Factor Pathway Inhibitor (TFPI) | Inactivates FXa/FVIIa/TF complex. Limits FX activation. |
| Activated Protein C (APC) | Inactivates FVa, FVIIIa. |
| Protein S | Cofactor for Protein C; inhibits prothrombinase complex. |
| Thrombin Activatable Fibrinolysis Inhibitor (TAFI) | Attenuates fibrinolysis; removes C-terminal lysine/arginine residues preventing plasminogen binding. |
Coagulation Cascade
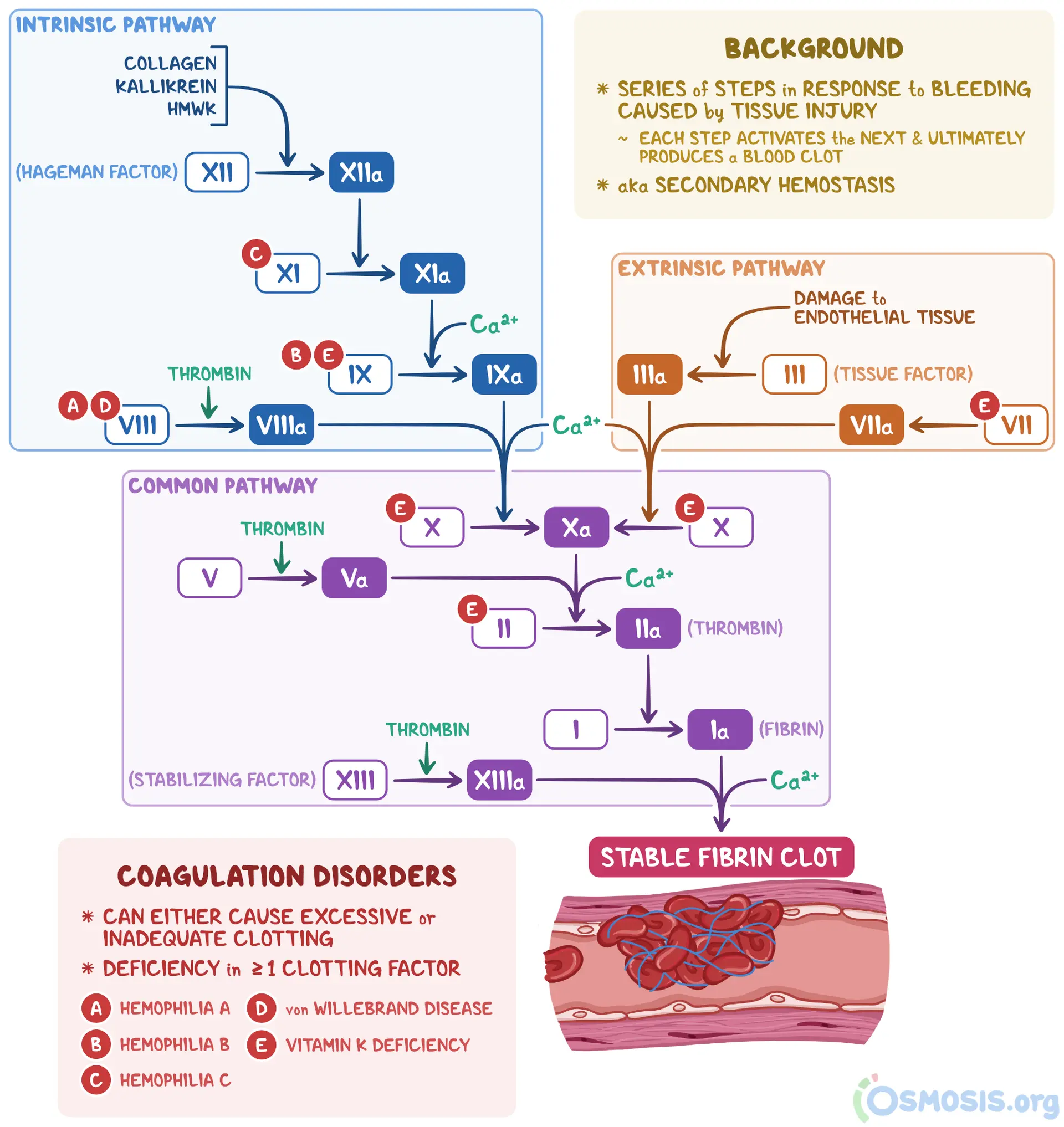 Source: Osmosis
Source: Osmosis
In Vivo Cell-Based Model Of Coagulation
Physiologic coagulation cascade occurs in three overlapping phases on cell surfaces.
Initiation Phase
- Occurs localized to tissue-factor bearing cells.
- Endothelial injury exposes subendothelial tissue factor (TF).
- Circulating Factor VII binds exposed TF; becomes rapidly activated.
- FVIIa/TF complex generates activated Factor X (FXa) and Factor IX (FIXa).
- FXa activates Factor V (FV).
- FXa complexes with FVa; generates initial small amounts of thrombin.
Amplification Phase
- Procoagulant stimulus transfers to activated platelet surface at injury site.
- Initial small thrombin amounts enhance platelet adhesion.
- Platelets become fully activated; internal membrane phospholipids (phosphatidylserine) externalize.
- Thrombin activates FV, Factor VIII (FVIII), and Factor XI (FXI).
- Thrombin unbinds FVIII from von Willebrand factor (vWF) carrier protein, enabling participation in cascade.
Propagation Phase
- Occurs on activated platelet phospholipid surface.
- “Tenase” complex (FIXa + FVIIIa + calcium + phospholipid) assembles on platelet surface.
- Tenase complex efficiently generates massive amounts of FXa.
- “Prothrombinase” complex (FXa + FVa + calcium + phospholipid) assembles on platelet surface.
- Prothrombinase complex efficiently converts prothrombin (FII) into massive thrombin (FIIa) burst.
- Thrombin cleaves fibrinopeptides A and B from fibrinogen (FI).
- Residual peptide chains aggregate via hydrogen bonds forming soluble fibrin monomers.
- Thrombin activates Factor XIII (FXIII).
- FXIIIa converts soluble fibrin monomers into stable cross-linked fibrin polymers.
- Forms definitive, stable fibrin-platelet hemostatic barrier.
In Vitro Classical Coagulation Cascade
Conceptual model dividing cascade into three pathways; useful for in vitro laboratory testing interpretation.
Extrinsic Pathway
- Serves as primary initiating pathway for coagulation.
- Measured by prothrombin time (PT).
- Initiated by tissue factor (thromboplastin) and calcium.
- Involves TF and FVII.
Intrinsic Pathway
- Functions as regulatory amplification loop.
- Measured by activated partial thromboplastin time (aPTT).
- Initiated by contact activation (glass, silica, ellagic acid).
- Involves high-molecular-weight kininogen, prekallikrein, FXII, FXI, FIX, FVIII.
Common Pathway
- Convergence point of intrinsic and extrinsic pathways.
- Involves FX, FV, FII (prothrombin), FI (fibrinogen).
- Culminates in fibrin clot formation.
Plasma Coagulation Factors Profile
All plasma coagulation factors synthesize primarily in liver. FVIII additionally produces in endothelial cells. Factors II, VII, IX, and X require vitamin K for posttranslational gamma-carboxylation.
| Factor | Common Name | Biologic Half-Life (Hours) | Inheritance Of Deficiency |
|---|---|---|---|
| I | Fibrinogen | 56-82 | Autosomal Recessive/Dominant |
| II | Prothrombin (Vitamin K dependent) | 45-60 | Autosomal Recessive |
| V | Proaccelerin, labile factor | 36 | Autosomal Recessive |
| VII | Proconvertin, stable factor (Vitamin K dependent) | 5 | Autosomal Recessive |
| VIII | Antihemophilic factor | 8-12 | X-linked Recessive |
| IX | Christmas factor (Vitamin K dependent) | 12-24 | X-linked Recessive |
| X | Stuart-Prower factor (Vitamin K dependent) | 24-60 | Autosomal Recessive |
| XI | Plasma thromboplastin antecedent | 48 | Autosomal Variable |
| XII | Hageman factor | 48-52 | Autosomal Dominant |
| XIII | Fibrin-stabilizing factor | 168-240 | Autosomal Recessive |
Natural Inhibitors Of Coagulation
Regulate extension of clotting process; maintain fluid state of blood; prevent systemic thrombosis.
| Inhibitor | Primary Target Substrates | Mechanism Of Action |
|---|---|---|
| Antithrombin (AT) | Thrombin, FXa, FIXa, FXIa, FXIIa | Serine protease inhibitor (SERPIN). Neutralizes procoagulants. Activity dramatically accelerated by heparin binding. |
| Tissue Factor Pathway Inhibitor (TFPI) | FXa/FVIIa/TF complex | Limits activation of FX by FVIIa and TF. Shifts activation site to FIX. |
| Protein C | FVa, FVIIIa | Vitamin K-dependent zymogen. Activated by thrombin-thrombomodulin complex. Proteolyzes activated cofactors. |
| Protein S | Prothrombinase complex, tenase complex | Vitamin K-dependent cofactor. Accelerates activated Protein C. Directly inhibits prothrombinase and tenase complexes. |
Fibrinolysis System
Provides physiologic mechanism for removal of deposited fibrin; reestablishes vascular integrity.
Activation Of Fibrinolysis
- Plasminogen incorporates into fibrin clot during thrombin formation.
- Tissue plasminogen activator (t-PA) serves as principal intravascular activator.
- t-PA binds fibrin via lysine-binding sites; markedly increases plasmin generation efficiency.
- Urokinase-type plasminogen activator (u-PA) activates plasminogen independent of fibrin.
- Plasmin (active enzyme) cleaves fibrin and fibrinogen into fibrin-degradation products (fragments X, Y, D, E).
- Fibrin split products feature heparin-like effects; inhibit platelet adhesion.
Inhibition Of Fibrinolysis
- Alpha-2-antiplasmin: Directly inhibits plasmin activity.
- Plasminogen Activator Inhibitor-1 (PAI-1): Primary physiologic inhibitor of t-PA.
- Thrombin Activatable Fibrinolysis Inhibitor (TAFI): Removes C-terminal lysine/arginine residues from fibrin. Prevents plasminogen binding; attenuates fibrinolysis.
Laboratory Evaluation Of Coagulation Cascade
Clinical evaluation utilizes specific screening tests to isolate pathway defects.
| Laboratory Finding | Associated Coagulation Defects |
|---|---|
| Isolated Prolonged PT | FVII deficiency, early vitamin K deficiency, early liver synthetic dysfunction. |
| Isolated Prolonged aPTT | Hemophilia A (FVIII), Hemophilia B (FIX), Hemophilia C (FXI), FXII deficiency. von Willebrand disease. Acquired FVIII/FIX/FXI inhibitors. Lupus anticoagulant. Heparin effect. |
| Prolonged PT and aPTT | Common pathway factor deficiency (FII, FV, FX). Severe fibrinogen deficiency. Disseminated intravascular coagulation (DIC). Severe liver disease. Marked vitamin K deficiency. Dilutional coagulopathy. |
| Normal PT/aPTT With Bleeding | FXIII deficiency. von Willebrand disease (mild). Platelet dysfunction. Mild specific factor deficiencies (FVIII, FIX, FXI). Alpha-2-antiplasmin deficiency. Collagen vascular diseases (Ehlers-Danlos). |
Advanced Coagulation Assays
- Mixing Studies: Differentiate factor deficiency (PT/aPTT corrects completely) from acquired circulating inhibitors (PT/aPTT fails to correct).
- Thrombin Time (TT): Assesses final conversion of fibrinogen to fibrin. Prolonged in hypofibrinogenemia, dysfibrinogenemia, heparin presence, or elevated fibrin split products.
- Specific Factor Assays: Quantify precise functional percentage of individual clotting proteins to definitively diagnose severity of hemophilias and rare bleeding disorders.