Syndromic stigmata: Evaluate for congenital bone marrow failure syndromes (Fanconi anemia, thrombocytopenia with absent radii).
Diagnostic Algorithm
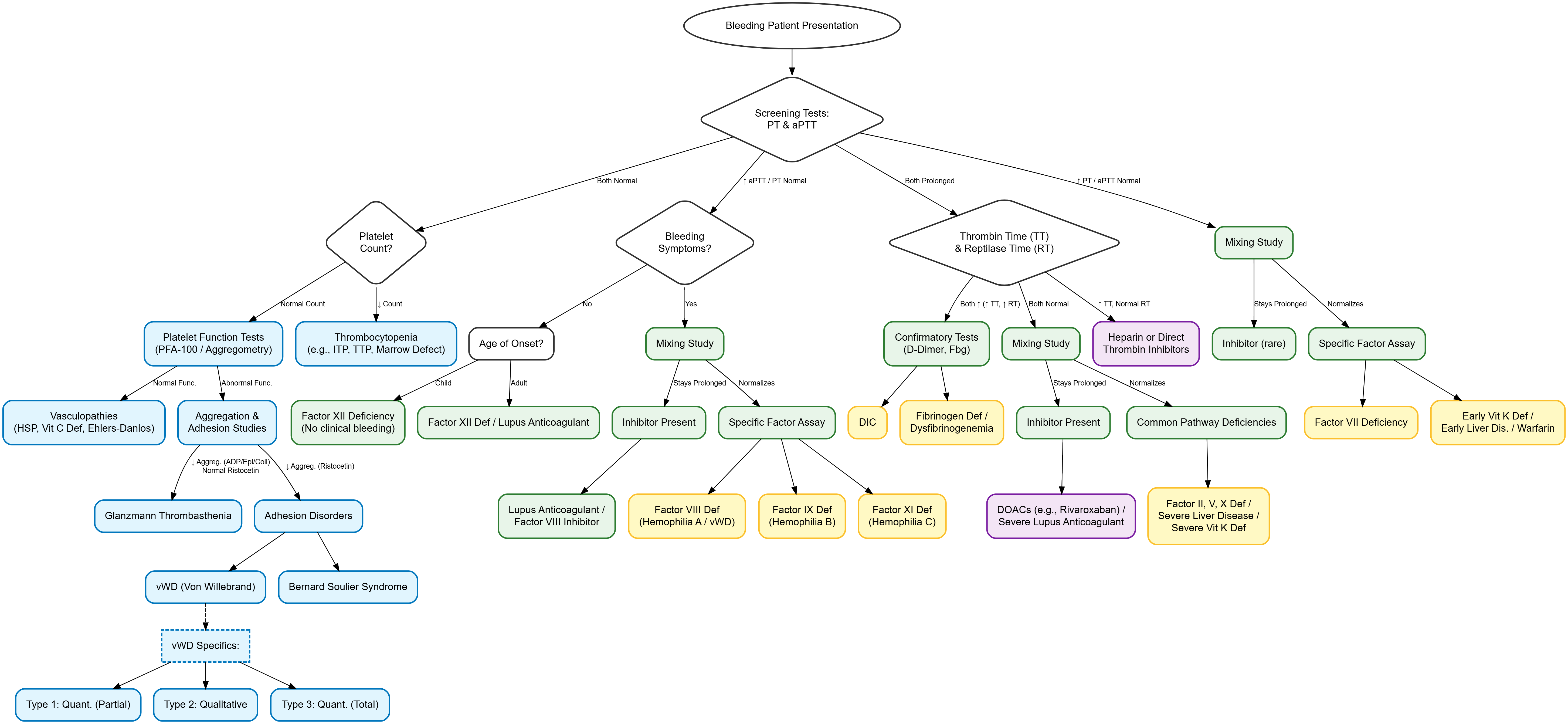
Interpretation Of Screening Tests
Laboratory Finding
Associated Etiologies
Isolated Prolonged PT
Factor VII deficiency, early vitamin K deficiency, early liver synthetic dysfunction.
Isolated Prolonged aPTT
Hemophilia A (FVIII), Hemophilia B (FIX), Hemophilia C (FXI), FXII deficiency, von Willebrand disease, acquired inhibitors, heparin effect, lupus anticoagulant.
Prolonged PT And aPTT
Common pathway factor deficiency (FII, FV, FX), afibrinogenemia, dysfibrinogenemia, disseminated intravascular coagulation (DIC), severe liver disease, marked vitamin K deficiency, dilutional coagulopathy.
Normal PT And aPTT
Platelet dysfunction, factor XIII deficiency, mild von Willebrand disease, vascular connective tissue defects (Ehlers-Danlos, scurvy).
Advanced Coagulation Laboratory Assessment
Mixing Studies
Differentiates factor deficiency from acquired circulating inhibitors.
Normal plasma added to patient plasma (1:1 ratio).
Failure to correct or partial correction indicates coagulation inhibitor (e.g., specific factor antibody, lupus anticoagulant).
Thrombin Time And Reptilase Time
Thrombin Time (TT): Measures final conversion of fibrinogen to fibrin. Prolonged by hypofibrinogenemia, dysfibrinogenemia, heparin, or elevated fibrin degradation products.
Reptilase Time: Utilizes snake venom to clot fibrinogen. Unaffected by heparin. Differentiates heparin contamination (prolonged TT, normal reptilase) from true fibrinogen defects (prolonged TT and reptilase time).
Specific Factor Assays
Quantify precise functional activity of individual clotting proteins.
Indicated when mixing studies normalize specific pathway abnormalities.
Essential for classifying severity of hemophilias (Severe <1%, Moderate 1-5%, Mild 5-40%).
Von Willebrand Disease Panel
vWF Antigen (vWF:Ag): Quantifies total vWF protein concentration.
Ristocetin Cofactor Activity (vWF:RCo): Assesses functional interaction between vWF and platelets.
Factor VIII Activity: Often reduced due to absent vWF chaperone function.
vWF Multimer Analysis: Visualizes molecular weight multimers. Identifies absence of high-molecular-weight multimers in Types 2A and 2B.
Global Hemostatic Tests
Thromboelastography (TEG): Assesses viscoelastic properties of whole blood clot formation under low shear.
Evaluates clot initiation (R time), kinetics (K time, alpha angle), strength (Maximum Amplitude), and fibrinolysis.
Manifest with deep tissue bleeding: hemarthroses, large intramuscular hematomas, delayed surgical bleeding.
Hemophilia A And B
Genetics: X-linked recessive. Hemophilia A (Factor VIII deficiency); Hemophilia B (Factor IX deficiency).
Laboratory: Prolonged aPTT, normal PT, normal platelet count. Specific factor assays establish definitive diagnosis.
Clinical: Severe disease (<1% factor) causes spontaneous joint and muscle bleeds. Target joints develop from recurrent hemarthroses.
Treatment: Specific recombinant factor concentrates. Prophylaxis recommended for severe phenotypes to prevent arthropathy.
Von Willebrand Disease
Genetics: Most common inherited bleeding disorder. Mostly autosomal dominant.
Pathophysiology: Deficient or defective vWF impairs platelet adhesion and factor VIII protection.
Classification:
Type 1: Partial quantitative deficiency.
Type 2: Qualitative functional defects (Subtypes 2A, 2B, 2M, 2N).
Type 3: Complete quantitative absence.
Treatment: Desmopressin (DDAVP) for Type 1. vWF-containing factor concentrates for severe types or unresponsiveness. Antifibrinolytics for mucosal bleeding.
Disseminated Intravascular Coagulation (DIC)
Acquired clinicopathologic syndrome.
Triggers include sepsis, trauma, malignancy, massive tissue injury.
Pathogenesis involves unregulated thrombin generation, widespread microvascular fibrin deposition, and consumption of platelets/coagulation factors.
Management: Treat underlying trigger. Replace depleted components (fresh frozen plasma, cryoprecipitate, platelets) only for active hemorrhage or invasive procedures.
Vitamin K Deficiency
Required for gamma-carboxylation of factors II, VII, IX, X, Protein C, Protein S.
Prolongs PT early (factor VII has shortest half-life), followed by aPTT.
Responsive to parenteral vitamin K administration.
Liver Disease Coagulopathy
Liver synthesizes most procoagulant and natural anticoagulant proteins.