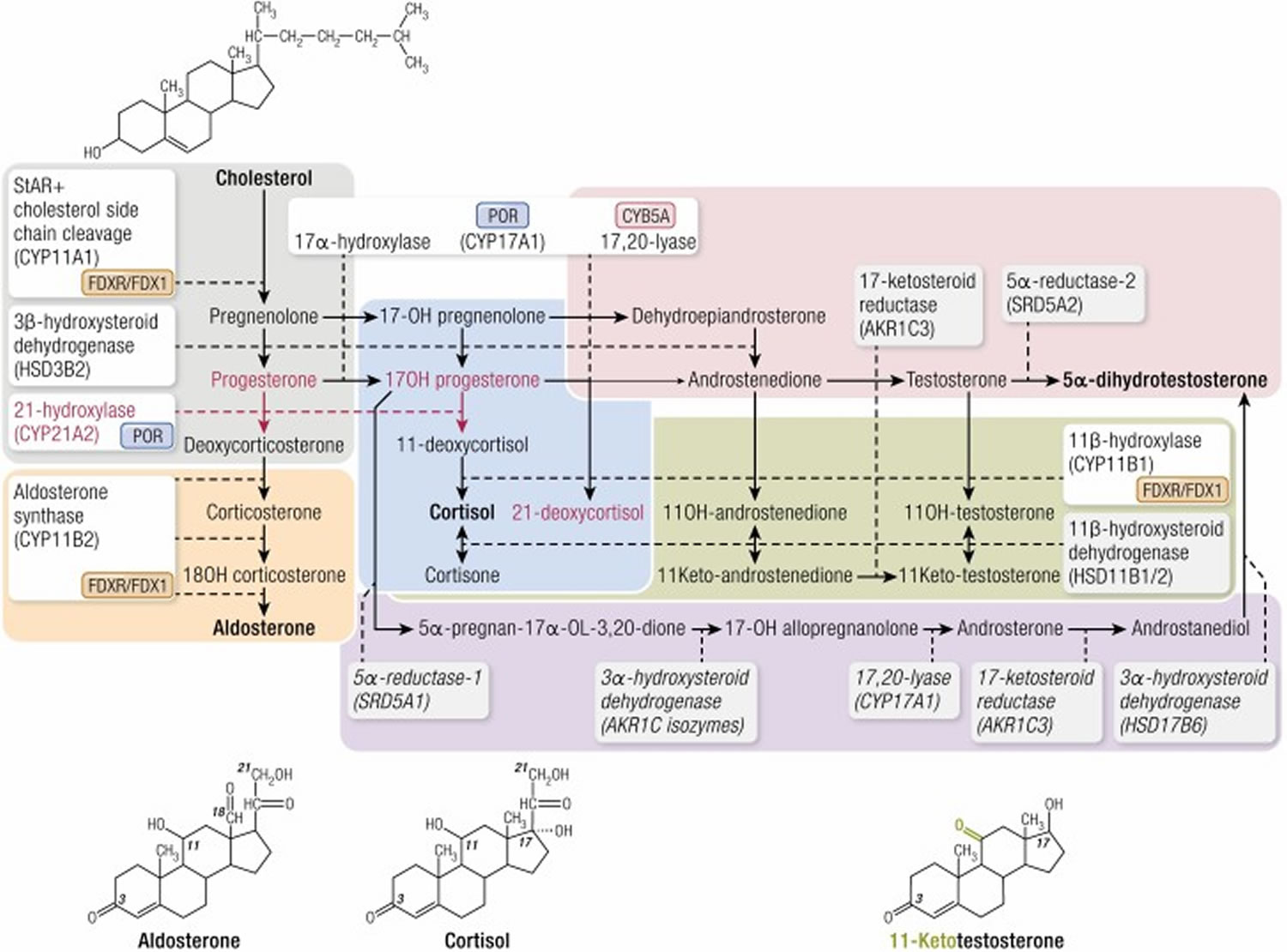
Congenital adrenal hyperplasia (CAH) encompasses a family of autosomal recessive disorders characterized by genetic defects in the enzymes required for cortisol biosynthesis.
The fundamental pathophysiology shared by these disorders involves insufficient cortisol production, which eliminates normal negative feedback inhibition on the hypothalamus and pituitary gland.
The resulting hypersecretion of corticotropin (ACTH) drives continuous stimulation of the adrenal cortex, leading to adrenocortical hyperplasia and the massive accumulation of intermediate steroid metabolites proximal to the enzymatic block.
Depending on the specific enzyme deficiency, these accumulated precursors are shunted into alternative open pathways, resulting in characteristic patterns of mineralocorticoid deficiency or excess, as well as varying degrees of androgen deficiency or excess.
Clinical phenotypes range from fatal neonatal salt-wasting crises and ambiguous genitalia to delayed puberty, hypertension, and asymptomatic nonclassic forms presenting in adulthood.
Summary Table of Congenital Adrenal Hyperplasia Types
Enzyme Deficiency
Gene (Locus)
Mineralocorticoid Effect
Genital Presentation (46,XX)
Genital Presentation (46,XY)
Key Diagnostic Biochemical Elevations
21-Hydroxylase
CYP21A2 (6p21.3)
Salt-wasting (low aldosterone) or normal
Ambiguous (virilized)
Normal male (postnatal virilization)
17-Hydroxyprogesterone (17-OHP), Androstenedione
11β-Hydroxylase
CYP11B1 (8q24.3)
Hypertension (high DOC)
Ambiguous (virilized)
Normal male (postnatal virilization)
11-Deoxycortisol, Deoxycorticosterone (DOC)
3β-Hydroxysteroid Dehydrogenase (Type 2)
HSD3B2 (1p12)
Salt-wasting
Ambiguous (mild virilization from DHEA)
Ambiguous (undervirilized)
17-Hydroxypregnenolone, DHEA, Pregnenolone
17α-Hydroxylase / 17,20-Lyase
CYP17A1 (10q24.3)
Hypertension (high DOC & corticosterone)
Normal female (delayed puberty/infantilism)
Ambiguous or normal female (undervirilized)
DOC, Corticosterone
Steroidogenic Acute Regulatory Protein (Lipoid CAH)
STAR (8p11.2)
Severe salt-wasting
Normal female (delayed puberty)
Normal female (undervirilized)
None (Low/absent levels of all steroid hormones)
Cholesterol Side-Chain Cleavage
CYP11A1 (15q23-24)
Severe salt-wasting
Normal female
Normal female or ambiguous
None (Low/absent levels of all steroid hormones)
P450 Oxidoreductase (POR)
POR (7q11.2)
Variable (often normal)
Ambiguous (virilized)
Ambiguous (undervirilized)
Variable; often mixed 17-OHP and progesterone elevation
21-Hydroxylase Deficiency (CYP21A2)
Epidemiology
Most common inborn error of steroidogenesis.
Causes >90% of Congenital Adrenal Hyperplasia (CAH) cases.
Glucocorticoids: Oral hydrocortisone in growing children (10-15 mg/m²/day) to suppress ACTH/androgen overproduction.
Mineralocorticoids: Fludrocortisone (0.05-0.15 mg/day; newborns may require up to 0.4 mg).
Sodium supplementation: 1-2 g NaCl/day required in newborns.
Surgical intervention: Genital reconstruction for affected 46,XX DSD females.
11β-Hydroxylase Deficiency (CYP11B1)
Overview & Epidemiology
Rare autosomal recessive endocrine disorder.
Represents 3% to 5% of Congenital Adrenal Hyperplasia (CAH) cases globally.
General population incidence: 1 in 100,000 to 1 in 250,000 live births.
Highest prevalence in specific ethnic isolates due to founder effects.
Epidemiological Feature
Details
Moroccan Jewish Population
- Incidence: 1 in 5,000 to 1 in 7,000 live births.- Predominant mutation: R448H (Arg448 to His).
Tunisian Arab Population
- Common mutations: Q356X, G379V.
Turkish Population
- Accounts for up to 13.5% of CAH cases.
Genetics & Molecular Biology
Genetic Feature
Details
Gene
- CYP11B1.
Locus
- Chromosome 8q21-22 (also cited as 8q24.3).
Inheritance
- Autosomal recessive.
Protein Encoded
- P450c11β (11β-hydroxylase).
Structural Homology
- 93% amino acid sequence identity with CYP11B2 (aldosterone synthase).
Gene Proximity
- Closely linked in tandem with CYP11B2 on chromosome 8.
Mutation Types
- Over 100 CYP11B1 mutations identified (missense, nonsense, deletions).
Pathophysiology of Steroidogenesis
Normal Enzyme Function
P450c11β resides in mitochondrial inner membrane of adrenal zona fasciculata and zona reticularis.
Catalyzes 11β-hydroxylation.
Converts 11-deoxycortisol to cortisol.
Converts 11-deoxycorticosterone (DOC) to corticosterone.
Acts on C-19 steroids: Converts androstenedione to 11β-hydroxyandrostenedione; testosterone to 11β-hydroxytestosterone (precursors to potent 11-oxygenated androgens).
Consequences of Enzymatic Blockade
Cortisol synthesis impaired → loss of negative feedback loop.
Corticotropin-releasing hormone (CRH) and Adrenocorticotropic hormone (ACTH) secretion excessively stimulated.
- Shunting of accumulated precursors into sex steroid biosynthesis pathways.- Elevated dehydroepiandrosterone (DHEA), androstenedione, and testosterone.- Potential utilization of alternate “backdoor pathway” converting excess 17-OHP to dihydrotestosterone (DHT) independently of testosterone.
Secondary 17-OHP Elevation
- Accumulated 11-deoxycortisol competitively inhibits P450c21 (21-hydroxylase).- Results in secondary elevation of 17-hydroxyprogesterone (17-OHP).
Clinical Manifestations
Classic 11β-Hydroxylase Deficiency
Age / Sex
Manifestations
46,XX Neonates (Females)
- Disorder of Sex Development (DSD) / Ambiguous genitalia.- Clitoromegaly, posterior labial fusion, urogenital sinus.- Internal Müllerian structures (uterus, fallopian tubes, upper vagina) preserved.
46,XY Neonates (Males)
- Normal male external genitalia at birth.- Isosexual precocity may develop postnatally.
Both Sexes (Neonatal)
- Transient mild salt loss/hyponatremia despite DOC excess.- Driven by physiological neonatal renal resistance to mineralocorticoids.- Resolves quickly; frequently causes early misdiagnosis as 21-hydroxylase deficiency.
Childhood (Both Sexes)
- Accelerated linear growth velocity.- Markedly advanced skeletal maturation (bone age).- Premature epiphyseal fusion → stunted final adult stature.- Premature pubarche, axillary hair, severe acne.- Deepening of voice, excessive muscular development.
Cardiovascular
- Hypertension develops in ~65% of patients.- Often delayed onset (takes years to manifest).- Chronic hypokalemia, polyuria, nocturia, muscle weakness.- Long-standing untreated cases risk ventricular hypertrophy, retinopathy, cerebrovascular events.
Bone Age Radiograph: Advanced skeletal maturation relative to chronologic age.
Pelvic Ultrasound: Confirms presence of Müllerian structures (uterus, ovaries) in virilized 46,XX neonates.
Differential Diagnosis
Disorder
Differentiating Clinical & Laboratory Features from 11β-OHD
21-Hydroxylase Deficiency
- Most common CAH (>90%).- Salt-wasting: Hyponatremia, hyperkalemia, hypotension (NO hypertension).- Massive elevation of 17-OHP; 11-deoxycortisol is LOW.- Elevated PRA.
17α-Hydroxylase Deficiency
- Hypertension and hypokalemia present (like 11β-OHD).- Deficient sex steroids → 46,XY undervirilized (phenotypic female/ambiguous); 46,XX sexual infantilism/primary amenorrhea (NO virilization).
3β-HSD Deficiency
- Ambiguous genitalia in BOTH 46,XX and 46,XY.- Elevated Δ5 steroids (pregnenolone, 17-hydroxypregnenolone, DHEA).- High Δ5:Δ4 steroid ratio.
P450 Oxidoreductase (POR) Deficiency
- Ambiguous genitalia in both sexes.- Mixed features of 21-OHD and 17α-OHD.- Associated with Antley-Bixler skeletal dysplasia (craniosynostosis, radiohumeral synostosis).
Management
Pharmacotherapy
Modality
Intervention & Rationale
Glucocorticoid Replacement
- Drug: Hydrocortisone (children) to minimize growth suppression.- Mechanism: Replaces deficient cortisol, restores negative feedback, suppresses excessive ACTH.- Effect: Halts overproduction of DOC (resolving hypertension) and androgens (halting progressive virilization and bone age advancement).- Dosing: 10-15 mg/m2/day divided in 3 doses. Stress dosing (2x-3x) required during illness/surgery.
Mineralocorticoid Therapy
- Generally NOT required in 11β-hydroxylase deficiency.- Exception: May be transiently required in young infants who develop paradoxical salt-loss immediately upon initiation of glucocorticoid therapy (due to sudden suppression of DOC before normal aldosterone axis recovers).
Antihypertensive Agents
- Required if hypertension is long-standing, severe, or fails to resolve promptly with glucocorticoid suppression of DOC.- Drug of choice: Calcium channel blockers, Potassium-sparing diuretics (spironolactone).
Surgical & Adjunctive Therapy
Feminizing Genitoplasty: Reconstructive surgery (clitoroplasty/clitoral recession, vaginoplasty) for severely virilized 46,XX infants. Timed via multidisciplinary consensus (pediatric urology/endocrinology/psychology).
Growth Optimization: Close monitoring of height velocity, weight, and serial bone ages. Prevent overtreatment (causes iatrogenic Cushing/growth arrest) and undertreatment (causes premature epiphyseal fusion).
Genetic Counseling: Autosomal recessive transmission. 25% recurrence risk in subsequent pregnancies. Carrier testing available for family members.
3β-Hydroxysteroid Dehydrogenase Type 2 Deficiency (HSD3B2)
Pathophysiology
Rare, autosomal recessive variant of Congenital Adrenal Hyperplasia (CAH).
Caused by loss-of-function mutations in HSD3B2 gene (chromosome 1p13.1).
Impairs conversion of Δ5 steroids to Δ4-ketosteroids (pregnenolone → progesterone; 17-hydroxypregnenolone → 17-OHP; DHEA → androstenedione).
Leads to complete or partial deficiency of glucocorticoids, mineralocorticoids, and sex steroids.
Triggers compensatory ACTH hypersecretion and massive accumulation of dehydroepiandrosterone (DHEA).
Clinical Manifestations
Phenotype
Key Features
46,XX (Females)
- Mild virilization and clitoromegaly.- Driven by peripheral conversion of massive DHEA to testosterone via unaffected HSD3B1 isoenzyme.- Normal internal female organs.
46,XY (Males)
- Undervirilization, varying degrees of hypospadias, bifid scrotum, cryptorchidism.- Caused by insufficient testicular testosterone synthesis.- Intact Wolffian duct structures.
Systemic (Both)
- Classic salt-wasting crisis in the newborn period (hypotension, hyponatremia, hyperkalemia) due to aldosterone deficiency.- Late-onset nonclassic forms present with premature pubarche or hirsutism.
Diagnosis
Steroid Profile: Marked elevation of baseline and ACTH-stimulated Δ5 steroids (pregnenolone, 17-hydroxypregnenolone, DHEA).
Ratios: Elevated Δ5 to Δ4 steroid ratios.
Pitfall: May mimic 21-hydroxylase deficiency newborn screens. Extraglandular HSD3B1 converts accumulated 17-hydroxypregnenolone to 17-OHP, yielding false-positive high 17-OHP levels.
Other Labs: Low cortisol, low aldosterone, elevated plasma renin activity (PRA), and elevated ACTH.
Management
Steroid Replacement: Lifelong glucocorticoid (hydrocortisone) and mineralocorticoid (fludrocortisone) replacement.
Electrolytes: Sodium chloride supplementation in infancy.
Surgical/Hormonal: Genital reconstructive surgery as needed. Testosterone replacement for incompletely virilized 46,XY males raised as males.
Accounts for less than 1% of CAH cases and is caused by mutations in CYP17A1 located on chromosome 10q24.3.
The CYP17A1 enzyme performs two distinct reactions: 17α-hydroxylation (essential for cortisol and sex steroids) and 17,20-lyase activity (essential for producing DHEA and androstenedione, the precursors to testosterone and estradiol).
The complete enzyme blockade eliminates cortisol and sex steroid production, shifting all steroidogenic precursors into the mineralocorticoid pathway (DOC and corticosterone).
Clinical Presentation
Hypertension: The unchecked accumulation of DOC causes severe hypertension and hypokalemic alkalosis with suppressed renin and aldosterone.
Sexual Infantilism / DSD: Because sex steroid synthesis is abolished in both the adrenals and gonads:
46,XY Infants: Present with completely female external genitalia or severe ambiguity (undervirilization) due to the absence of fetal testosterone.
46,XX Infants: Present with normal female external genitalia at birth but completely fail to undergo puberty (primary amenorrhea and absent secondary sexual characteristics) due to the lack of estrogen production.
Diagnosis
Laboratory testing reveals elevated levels of DOC and corticosterone, alongside suppressed levels of 17-OHP, cortisol, androgens, and estrogens. Gonadotropins (LH and FSH) are elevated in pubertal-aged patients.
Congenital Lipoid Adrenal Hyperplasia
Pathophysiology and Genetics
This represents the most profound and severe form of CAH, leading to the disruption of all steroidogenesis (glucocorticoids, mineralocorticoids, and sex steroids).
It is primarily caused by mutations in the STAR gene (8p11.2), which encodes the Steroidogenic Acute Regulatory protein required to transport free cholesterol from the outer to the inner mitochondrial membrane.
The Two-Hit Model: In early gestation, StAR-independent pathways allow for trace steroidogenesis (approximately 14% of normal). The resulting cortisol deficiency triggers massive ACTH release, driving extreme cellular uptake of cholesterol. The accumulating cholesterol esters physically destroy the cellular architecture and induce chemical toxicity (auto-oxidation), irreversibly obliterating all steroidogenic capacity (the “second hit”).
Alternatively, an identical phenotype is caused by mutations in CYP11A1 (P450 side-chain cleavage enzyme), which prevents the direct conversion of cholesterol into pregnenolone.
Clinical Presentation
Infants typically present with severe, life-threatening acute adrenal crisis and profound salt-wasting shortly after birth.
Due to the destruction of fetal Leydig cells or the inability to synthesize testosterone, affected 46,XY genetic males are born with entirely female external genitalia and a blind vaginal pouch.
Müllerian structures (uterus, tubes) are absent in 46,XY patients because the undamaged Sertoli cells continue to secrete AMH.
In STAR mutations, the adrenal glands are massively enlarged and engorged with lipids; conversely, in CYP11A1 mutations, the adrenal glands appear normal or small on ultrasound.
Cytochrome P450 Oxidoreductase (POR) Deficiency
Pathophysiology and Genetics
A complex and relatively recently defined variant of CAH caused by mutations in the POR gene on chromosome 7q11.2.
POR is an essential flavoprotein that donates electrons to all microsomal cytochrome P450 enzymes, directly impacting the function of 21-hydroxylase, 17α-hydroxylase, and aromatase.
The varied impairment across multiple enzymes creates a uniquely mixed biochemical and clinical profile.
Clinical Presentation
Genital Ambiguity: Manifests in both genetic sexes. 46,XX females are virilized (partly due to the utilization of the alternative backdoor pathway to DHT), while 46,XY males are undervirilized due to combined 17α-hydroxylase/17,20-lyase impairment.
Maternal Virilization: Pregnant mothers carrying an affected fetus may experience progressive virilization during gestation because the mutant placental aromatase fails to convert abundant fetal androgens into estrogens.
Skeletal Dysplasia: POR deficiency is strongly associated with the Antley-Bixler syndrome phenotype, characterized by severe skeletal anomalies including craniosynostosis, midface hypoplasia, radiohumeral synostosis, and bowing of the femora.
Management of Congenital Adrenal Hyperplasia
Pharmacological Therapy
Glucocorticoid Replacement: The primary goal is to replace deficient cortisol and suppress the excessive ACTH drive, thereby mitigating adrenal androgen overproduction. Oral hydrocortisone is the preferred therapy in growing children to minimize the risk of growth suppression.
Mineralocorticoid Replacement: Patients with salt-wasting forms strictly require fludrocortisone to maintain sodium balance, prevent hyperkalemia, and normalize plasma renin activity.
Sodium Supplementation: Infants require oral sodium chloride supplementation because the infant diet (breast milk or formula) is naturally low in sodium.
Surgical Considerations and Gender Rearing
Decisions regarding the sex of rearing and the timing of genitoplasty in severely virilized 46,XX infants are complex and require a multidisciplinary approach encompassing endocrinologists, urologists, and psychologists.
46,XX individuals with CAH possess normal internal reproductive organs (ovaries and uterus) and retain full potential for future fertility; the vast majority are raised as females and identify with the female gender.
Surgical correction (e.g., clitoroplasty and vaginoplasty) is highly individualized, and current paradigms weigh the benefits of early anatomical correction against the risks of long-term sexual dysfunction and the child’s future autonomy.
Long-Term Monitoring
The adequacy of therapy must be routinely monitored by tracking the child’s linear growth velocity and skeletal maturation (bone age).
Biochemical monitoring focuses on finding a balance: measuring androstenedione and testosterone to ensure adequate androgen suppression, while using 17-OHP to guard against the overtreatment and resultant iatrogenic Cushing syndrome.
Plasma renin activity (PRA) is monitored to gauge the precise adequacy of mineralocorticoid and sodium replacement.
In adolescent and adult males with CAH, routine testicular ultrasound screening is mandated to detect testicular adrenal rest tumors (TARTs), which are driven by chronic ACTH excess and can cause irreversible infertility.